
RDKitにおける記述子の扱い方をリピンスキーの法則を通して学ぶ
pythonでケモインフォマティクスを行う際の定番であるRDKitについて,これまで4つのエントリーに分けて基礎から使い方を説明してきました.
これまではRDKit上でどのように分子を構築し取り扱うかに集中して学んできたと言えます.ここからは,分子の性質をどのように表現・計算するかについて学びます.ようやくインフォマティクス・分析らしい要素が出てきます.
今回は分子の性質を表現する「記述子」について,RDKitではどのように扱うかをリピンスキーのルール・オブ・ファイブを題材として学んでいきます.
分子の準備
今回も「Platinum dataset」を使って分析を行います.このデータセットは小さい割に,それなりに構造多様性に富み,ドラッグライクな性質を持っている低分子に関するデータセットですので扱いやすいと言えます.CheEMBLなどのデータセットはもちろん有用なのですが,数が多すぎますので練習には向かないと考えています.
さてまずは必要なライブラリのimportを行いましょう.
# ライブラリのimport
from rdkit import rdBase, Chem
from rdkit.Chem import AllChem, Descriptors
print(rdBase.rdkitVersion) # 2020.09.3
import pandas as pd
import seaborn as sns
# 分子の読み込み
suppl = Chem.SDMolSupplier('./platinum_dataset_2017_01.sdf')
mols = [x for x in suppl if x is not None]
print(len(mols)) # 4548
ケモインフォマティクスにおける記述子とは何か?
記述子(デスクリプター)とは分子の性質を決める指標のことです.一口に性質といっても,水溶性や大きさももちろん性質ですし,生理活性の有無なども性質といえるかもしれません.特にケモインフォマティクスの分野では分子の構造から導くことが可能なものを記述子と呼んでいます.性質を記述するものということです.
多くの場合は,複数の記述子の組み合わせによって生理活性などの実験データを理解し,最終的には予測するモデル(QSARモデル)を作ることが記述子を用いる目的になります.これまで非常に多くの記述子が開発・提唱されてきましたし,これからもされ続けると思います.
既にケモインフォマティクスにおける記述子をまとめた分厚い本ができあがるほどに多くの種類がありますので,開発された記述子の全てを覚えることは不可能ですし,意味もないでしょう.合成化学者としては,LogPやTPSAなどのよく使うものについて理解を深めておくことが大切だと思います.
RDKitで記述子を計算する
RDKitにおける記述子はその性質や出典によって色々なモジュールに散在していますが,Chem.Descriptorsがほぼ全てをカバーしていますので,今回はこちらをimportとして説明していきます.
RDKitで計算可能な記述子はdesclistまたは_desclistに保存されていて,その数は2020.09のアップデート段階で208にもなります.すごい数にも思えますが,これでも開発されてきた記述子のほんの一部です.
desc_list = Descriptors.descList
print(len(desc_list)) # 208
for i in desc_list[:3]:
print(i)
('MaxEStateIndex', function MaxEStateIndex at 0x13ac600d0)
('MinEStateIndex', function MinEStateIndex at 0x13ac60160)
('MaxAbsEStateIndex', function MaxAbsEStateIndex at 0x13ac601f0)
descListの中身は(記述の名前,記述子を求める関数)を集めたリストになっています.そのため計算したい記述子の名前がわかっていれば対になっている計算関数を呼び出すだけで記述子の値が取得できます.下の表にも代表的なものをまとめておきますが,基本的にはNum’XXX’といった‘XXX’の数を数える記述子が大半だと考えて大丈夫です.
「The RDKit documentation:List of Available Descriptors
「Package rdkit :: Package Chem :: Module Descriptors」
| 記述子の名前 | 説明 |
|---|---|
| MolLogP | Crippenらによる原子ベースのLogP指標 |
| MolWt | 分子量 |
| NumHAcceptors | 水素結合アクセプターの数 |
| NumHDonors | 水素結合ドナーの数 |
| NumHeteroatoms | へテロ元素の数 |
| NumRotatableBonds | 回転可能な結合数 |
| FractionCSP3 | 全炭素数におけるsp3炭素の割合 |
| TPSA | トポロジカル極性表面積 |
具体例:リピンスキーの「ルール・オブ・ファイブ」
Lipinskiらが医薬品候補化合物の性質を調べる中で,経口医薬品になりやすい化合物の特徴としてまとめたものを「ルール・オブ・ファイブ(Rule of Five, RO5)」といいます.企業の創薬化学者はもちろん,アカデミアの研究者や学生でも聞いたことのある有名すぎるルールです.実際の運用に関してはいろいろと議論もありますが,感覚的に理解しやすく,また覚えやすいことからこれだけ広く受け入れられてきたと言えます.
ルール・オブ・ファイブの内容は以下の通りです.見ておわかり頂けるように,「5」を基調としたルールになっています.
- 水素結合ドナー(OH,NHの数)が5個以下であること
- 水素結合アクセプター(O,N原子の数)が10個以下であること
- LogPが5以下であること
- 分子量が500以下であること
RDKitにおけるリピンスキー記述子
Chem.rdMolDescriptors.CalcNumLipinskiHBD(mol)
Chem.rdMolDescriptors.CalcNumHBA(mol)
Chem.rdMolDescriptors.CalcNumHBD(mol)
なおLipinskiによる数え方では,水素結合アクセプターの数を酸素・窒素原子の個数によってカウントします.すなわち,N-メチルアセトアミドを考えてみると,水素結合アクセプターの数は2となります(カルボニル酸素とアミド窒素).一方で化学者が普通の感覚で数えると1つになるでしょう(カルボニル酸素のみ).
このような差異があるため,RDKitではChem.rdMolDescriptorsモジュールにオリジナルのLipinskiの数え方に基づいたカウントを行うメソッド
- CalcNumLipinskiHBA
- CalcNumLipinskiHBD
が実装されています.本来であれば以下のコードはこちらのメソッドを用いて実装を行うべきですが,今回はAPIの使い方の習得に重点を置いていますので,Chem.Descriptorsモジュールにある
- CalcNumHBA
- CalcNumHBD
という通常のカウント方法で計算するメソッドを使っています
今回はこのリピンスキーの法則についてRDKitを使って,化合物ごとに判定を行い,当てはまらなかった分子に対して簡単に分析を行ってみましょう.
まずはルール適用に必要な記述子である
- 水素結合ドナー
- 水素結合アクセプター
- Logp
- 分子量
を計算します.
オクタノール/水の分配係数であるLogP自体は実験値ですが,構造式から推定する方法がいくつも提唱されています(CLogP,ALogPなど).RDKitにはCrippenらによる原子ベースの方法を基にしたMolLogPが実装されていますのでそちらを使うことにします.
RDKitにおける記述子の計算方法
それでは具体的に計算していくこととします.
# 記述子の計算オブジェクト
calc = {}
for i,j in desc_list:
calc[i] = j
lipinski_list = ['NumHDonors', 'NumHAcceptors', 'MolWt', 'MolLogP']
# Ro5を判定する関数たち
def calc_lipinsk(mol):
lipinski = {}
for desc in lipinski_list:
lipinski[desc] = calc[desc](mol)
return lipinski
def check_lipinski(dic):
if dic['MolWt'] <= 500 and dic['MolLogP'] <= 5 and dic['NumHDonors'] <=5 and dic['NumHAcceptors'] <=10:
return True
else:
return False
def rule_of_five(mol):
prop = calc_lipinsk(mol)
if check_lipinski(prop):
return mol
# 具体的に計算
lipinski_mols = []
bad_mols = []
for m in mols:
if rule_of_five(m):
lipinski_mols.append(m)
else:
bad_mols.append(m)
len(lipinski_mols), len(bad_mols)
# (3574, 974)
データセットの中でリピンスキールールに適合するものが3574個,しないものが974個であることがわかりました.繰り返しになりますが,本来のLipinskiの数え方を用いた場合には,おそらくさらに多くの分子が適合しないであろうことを理解してください.
さて適合しない分子のうち最初の12個の構造をpy3Dmolを使って可視化してみましょう.
import py3Dmol
v = py3Dmol.view(width=800, height=600, linked=False, viewergrid=(3,4))
grid = [(i,j) for i in range(3) for j in range(4)]
for m, g in zip(bad_mols[:12], grid):
mb = Chem.MolToMolBlock(m)
v.addModel(mb, 'sdf', viewer=g)
v.setStyle({'stick': {}})
v.setBackgroundColor('#EEEEEE')
v.zoomTo()
v.show()

適合しない分子についてpandasのデータフレームに記述子の値をセットしてもう少し眺めてみることにしましょう.
NumHDonors = []
NumHAcceptors = []
MolWt = []
MolLogP = []
for m in bad_mols:
prop = calc_lipinsk(m)
NumHDonors.append(prop['NumHDonors'])
NumHAcceptors.append(prop['NumHAcceptors'])
MolWt.append(prop['MolWt'])
MolLogP.append(prop['MolLogP'])
df = pd.DataFrame({'NumHDonors': NumHDonors,
'NumHAcceptors': NumHAcceptors,
'MolWt': MolWt,
'MolLogP': MolLogP})
df.describe().round(2)
df.corr().round(2)
sns.pairplot(df, size=4)
bins = range(0,15,3)
df['NumHDonors_cut'] = pd.cut(df.NumHDonors, bins)
df.groupby('NumHDonors_cut')[['MolLogP','MolWt','NumHAcceptors']].mean().round(2)
基本的な統計量を眺めてみると,MolLogPはほとんど問題がなく,分子量ではじかれた分子が多そうに見えます.また水素結合アクセプターの数は比較的広く分散しているのに対し,水素結合ドナーの数はごく一部の分子が非常に大きいだけに見えます.
| MolLogP | MolWt | NumHAcceptors | NumHDonors | |
|---|---|---|---|---|
| count | 974.00 | 974.00 | 974.00 | 974.00 |
| mean | 0.98 | 481.71 | 8.24 | 3.24 |
| std | 4.33 | 84.27 | 4.19 | 2.15 |
| min | -10.16 | 182.17 | 0.00 | 0.00 |
| 25% | -3.12 | 424.43 | 5.00 | 2.00 |
| 50% | 2.21 | 506.08 | 7.00 | 3.00 |
| 75% | 5.04 | 546.63 | 11.00 | 4.00 |
| max | 8.84 | 600.67 | 21.00 | 12.00 |
水素結合ドナー・アクセプターの数がMolLogPと負の相関があることは予想通りですが,分子量が大きいからといってLogPへの影響は少ないようです.
| MolLogP | MolWt | NumHAcceptors | NumHDonors | |
|---|---|---|---|---|
| MolLogP | 1.00 | 0.30 | -0.77 | -0.65 |
| MolWt | 0.30 | 1.00 | 0.03 | -0.21 |
| NumHAcceptors | -0.77 | 0.03 | 1.00 | 0.36 |
| NumHDonors | -0.65 | -0.21 | 0.36 | 1.00 |
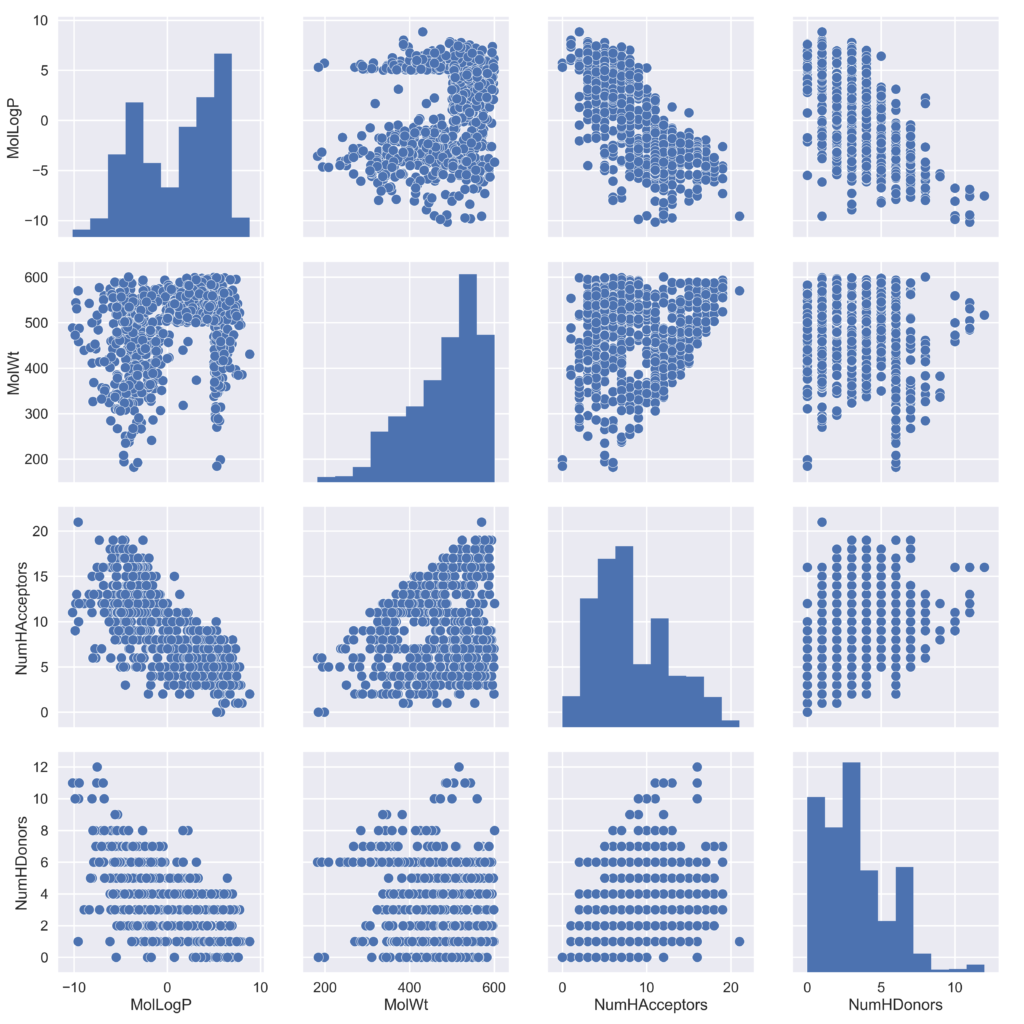
pandasのgroupby機能を使って水素結合ドナーの数で整理してみましたが,特に傾向は見えてこないようです.
| NumHDonors_cut | MolLogP | MolWt | NumHAcceptors |
|---|---|---|---|
| (0, 3] | 2.47 | 496.58 | 7.52 |
| (3, 6] | -1.38 | 470.45 | 9.69 |
| (6, 9] | -4.43 | 402.60 | 10.54 |
| (9, 12] | -8.49 | 506.21 | 13.15 |
今回はこのあたりで止めておきますが,記述子の計算を行うことがデータ分析の第一歩になることが理解できたと思います.一端化合物を用いたデータ分析の入り口に立つと,pandasの使い方についてさらに学んだり,統計学・機械学習を学習するモチベーションに繋がるのではないでしょうか.
終わりに
今回はこれまで構築したRDKitのおける分子であるMolオブジェクトを用いて,そこから得られる記述子について概説しました.記述子を組み合わせることで分子の性質を表現することが可能になりますから,化学構造(データ)から情報を得る第一歩を踏み出したと言っていいと思います.
今回取り扱ったの記述子は構造式中の特定構造の数を数えるなど,比較的古典的なものが中心でした.次回は特に3次元構造に特化した記述子を学んでいきたいと思います.昨今の医薬業界においてますます重要視されてきている指標ですので,RDKitを用いてしっかりと扱えるようにしていきましょう.
>>次の記事:「RDKitで立体的な分子の形を記述する」
コメント