
ケモインフォマティクスとは化学情報学とも呼ばれる分野で,コンピュータ・情報科学を用いて化学上の問題を取り扱う学問領域になります.そのためにはコンピュータで化合物の構造・性質などを取り扱う必要がありますが,人間とコンピュータでは化合物の認識方法が異なってきますので,専用のソフトウェアを用いていくことになります.
今回取り上げる「RDKit」は,代表的なケモインフォマティクス用のソフトウェアです.
- オープンソースのライブラリで開発が非常に活発
- 高価なソフトウェアに劣らない機能が無料で利用可能
- 初学者にも優しいプログラミング言語であるpythonを使って動かすことができる
などといった理由から幅広い支持を集めています.
本サイトでは,いくつかの記事を通してRDKitの使い方を基本から解説していきたいと思います.
RDKitとは
RDKitとはコンピュータで化合物情報を扱うケモインフォマティクス分野で用いられる代表的なオープンソースのライブラリです.pythonで扱うことができますが,多くのコードがC++で書かれていますので,実行速度も問題ありません.
開発が非常に盛んで,年に2回(3/4月と9/10月)のアップデートが行われています.またユーザー数も多く,ドキュメントやチュートリアルを扱ったJupyter Notebookも(英語なら)充実しています.メーリングリストでは基本的なエラーの報告や対処法から,高度な話題まで活発に議論されています.
これらの理由から,pythonで化合物を扱うなら是非とも習熟したいライブラリだと言えます.
- RDKitの公式サイトは「RDKit: Open-Source Cheminformatics Software」です.多くの資料やチュートリアルへのリンクが貼られています.
- 公式ドキュメントは「The RDKit Documentation」です.こちらを読みながら動かしていけば,基本的な操作はすぐにわかるようになると思います.
- 有志の方によるドキュメントの日本語訳もできました.
- メーリングリストの登録や過去ログは「SOURCE FORGE」から行います.やや流れるメール数が多いですが,RDKitをこれから本格的に使っていきたいと考えている方は「rdkit-discuss」の購読をおすすめします.
それでは具体的にインストール方法から順番に見ていきたいと思います.

RDKitのインストールと使い方
condaを利用する方法
RDKitのインストール方法はいくつかありますが,「The RDKit documentation:Installation」という公式のインストールガイドに記載がある通りに,新しいconda環境を作ってインストールするのが簡単で,推奨されています.下のコード例の「your-rdkit」の部分は作成する環境の名前になりますので,好きな名前を付けてください.
下の例ではpythonのバージョンを一緒に指定しています.RDKitだけを使うならあまり気にする必要はありませんが,TensorflowやPyTorchなどの他のライブラリと一緒に使いたい場合にpythonのバージョンが重要になる場合が多くあります.
conda create -c conda-forge -n your-rdkit python=3.9 rdkit
RDKitをインストールしたあとは新しく作成した環境(your-rdkit)に,
- numpy
- scipy
- pandas
- matplotlib
- sklearn
をはじめとした必要なライブラリをインストールしておきましょう.
作成した環境で下のコード例のようにrdkitが無事にimportできればインストールは成功です!(表示されるバージョンはみなさんの環境によって異なります)
import rdkit
print('rdkit version: ', rdkit.__version__)
# rdkit version: 2022.03.3
pipを利用する方法
上述のようにanacondaリポジトリの有償化や,そもそもcondaがpythonの標準化の流れと適合していないことなどから,なるべくcondaの利用を回避したい方も多いと思います.
condaはpython以外の多くのソフトウェアの配布にも使われており,管理人はconda以外では自分でビルドするしかインストール方法がない計算化学系のソフトを複数利用しているためconda環境にロックインされていますが,多くのpythonユーザーにとってはpipでインストールするほうが現実的だと思います.
RDKitは「RDKit Blog: Why the RDKit isn’t available on PyPi」というブログ記事でも説明されている通り,長らくpipではインストールできませんでした.ところがChristopher Künneth氏がwheelの作成に成功しrdkit-pypiという名前で2年ほど運用されてきました.この度,2022年6月にrdkitと名称変更されたことから,
pip install rdkit
というシンプルなコマンドでインストールが可能になりました.
brewを利用する方法
上述のようにpipを用いてインストールが可能になったため,ありがたみが薄れてしまいましたがhomebrewを使うことでMacやLinux上にRDKitをインストール可能です.
brew install rdkit
いずれかの方法でインストールが無事に成功したら,次は具体的にRDKitの使い方をみていきましょう.
RDKitで分子の読み込み
RDKitでは分子はMolオブジェクトとして扱われます.このオブジェクトは「SMILES」や「MOLファイル」といった一般的によく使われる形式から作成可能です.また逆にMolオブジェクトからこれらの形式への書き出しも可能です.
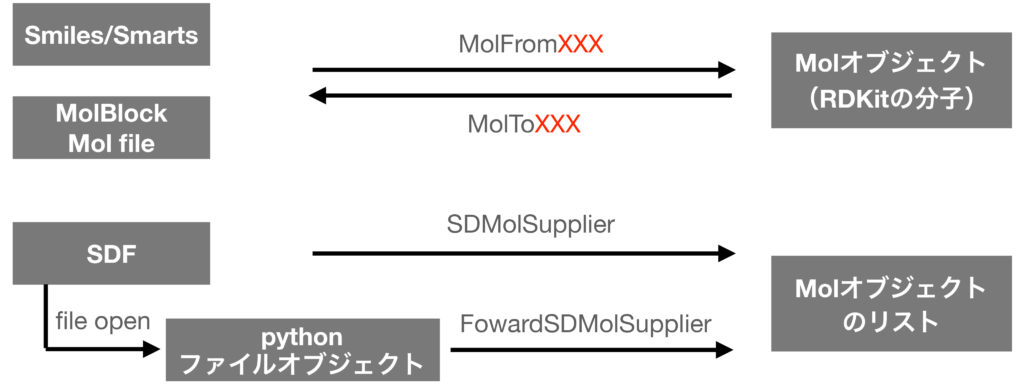
1分子の読み込み
上の図に全体の流れを簡単に示してありますが,
- 1分子を読み込む場合:Chem.MolFromXXX
- 1分子を書き込む場合:ChemMolToXXX
というメソッドを使います.XXXの部分をSmiles等に置き換えることで対応するフォーマットを指定することになります.
下のコードでは,まず「pythonで化合物データベースPubChemを使いこなす」という記事の復習を兼ねて,
- PubChemからベンゼンの検索を行いSmiles表記を取得
- その情報を用いてベンゼンのMolオブジェクトを作成
- 得られたオブジェクトをMOLブロックとして書き出し
という一連の作業を行っています.
import pubchempy as pcp
from rdkit import Chem
benzene = pcp.get_compounds('benzene', 'name')
if len(benzene) == 1: benzene = benzene[0]
smiles = benzene.canonical_smiles
print(smiles) # 'C1=CC=CC=C1'
mol_ben = Chem.MolFromSmiles(smiles)
print(type(mol_ben)) #
print(Chem.MolToMolBlock(mol_ben))
RDKit 2D
6 6 0 0 0 0 0 0 0 0999 V2000
1.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7500 -1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.7500 -1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.7500 1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7500 1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0
2 3 1 0
3 4 2 0
4 5 1 0
5 6 2 0
6 1 1 0
M END
試しにincludeStereo=Falseにして出力してみます.
print(Chem.MolToMolBlock(mol_ben, includeStereo=False))
座標が全て0になっているのが確認できます.
RDKit
6 6 0 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0
2 3 1 0
3 4 2 0
4 5 1 0
5 6 2 0
6 1 1 0
M END
複数分子の読み込み
RDKitでは,複数の分子をまとめる形式として一般的なSDFを読み込むことが可能です.
SDFとしては今回は下の図のようにfluorochemのページでサリチル酸誘導体を検索した結果を用いて,実際に複数分子を読み込んでみたいと思います.トップページ上部のメニューから「Products > Structure Search」へ進み構造検索を行います.「Search Type」を「Sub-Structure」に変更することで部分構造検索が行えます.431件のヒットに対して,「Export to Structure File」を選択することでSDFがゲットできます.
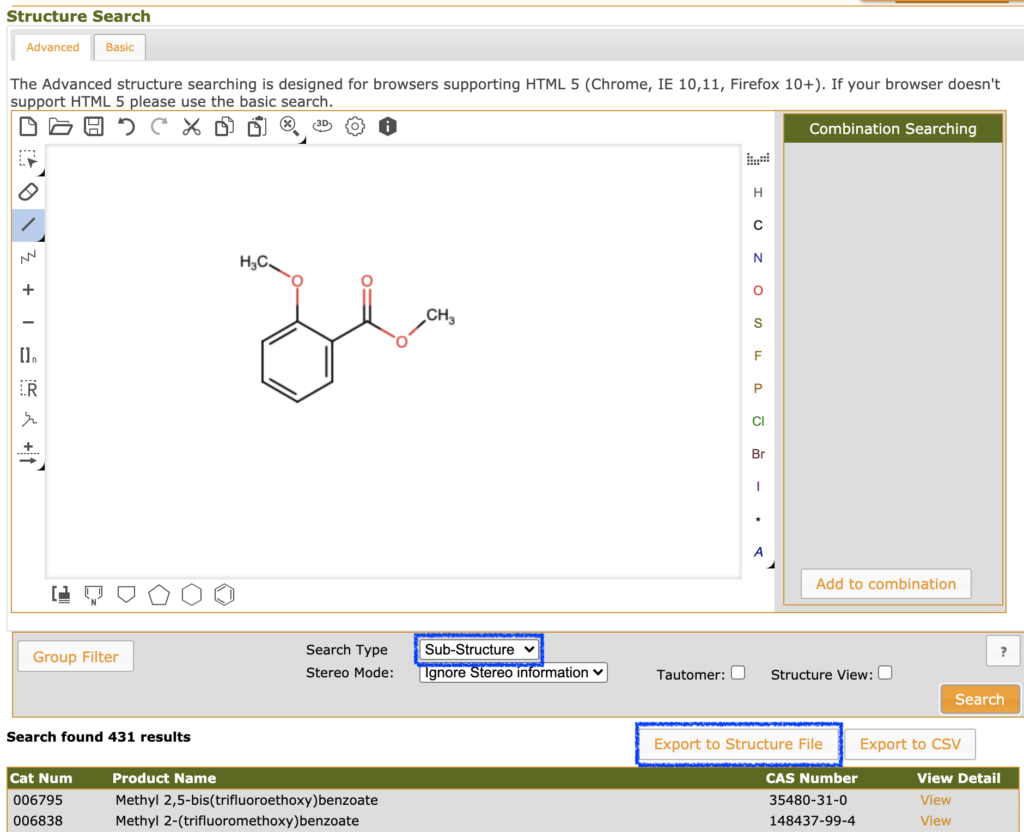
SDMolSupplierの利用
SDFからの読み込みにはSDMolSupplierを使うのが便利です.この際にRDKitは入力構造に何らかの不具合があって読み込めなかった場合はNoneを返すため,実際に分子に対して作業を開始する前にきちんと分子が読み込めているか確認するのが大切です.
また標準では水素原子を読み込みませんが(removeHs=True),下のコード例ではこれを無効にしています.今回の場合は1つのエラーもなく,全部で431個の分子を含むリストができあがりました.
suppl = Chem.SDMolSupplier('./salicylate_jan_2021.sdf', removeHs=False)
mols = [mol for mol in suppl if mol is not None]
print(len(mols)) # 431
pythonファイルオブジェクトの利用
RDKitにはファイルオブジェクトを読み込むことの出来るメソッドも用意されています.
下のコード例では先ほどと同じSDFを読んでいますが,ファイルオブジェクトを利用する方法ではgzip等で圧縮されたSDFもそのまま扱うことが可能な点が異なります.
同じファイルを使っていますので,こちらの方法でも431個の分子を含むリストができました.
with open('./salicylate_jan_2021.sdf', 'rb') as f:
fsuppl = Chem.ForwardSDMolSupplier(f)
fmols = [mol for mol in fsuppl if mol is not None]
print(len(fmols))
# 431
SDMolSupplierとForwardSDMolSupplierのもう1つの違いは,前者はMolオブジェクトのリストを作成するのに対し,後者は異なるという点が挙げられます.そのため上の例ではsuppl[0]とすると1つめのMolオブジェクトにアクセス可能ですが,fsuppl[0]ではエラーを返します.
SDFのpandasデータフレームへの読み込み
SDFとMOLファイルの違いとしては,
- 複数分子の構造を1つのファイルにまとめることができる
- SDFには分子構造だけでなくプロパティと呼ばれる情報を追加できる
といった点が挙げられます.
SDFからMolオブジェクトを作成しながら,設定されているプロパティをpandasのデータフレームとして一気に読み込むことができる方法がここで紹介する方法になります.SDFにたくさんのプロパティが設定されていて,その値を後で使いたい場合には非常に有用になります.
from rdkit.Chem import PandasTools
from IPython.display import HTML
df = PandasTools.LoadSDF('./salicylate_jan_2021.sdf')
HTML(df.head().to_html())
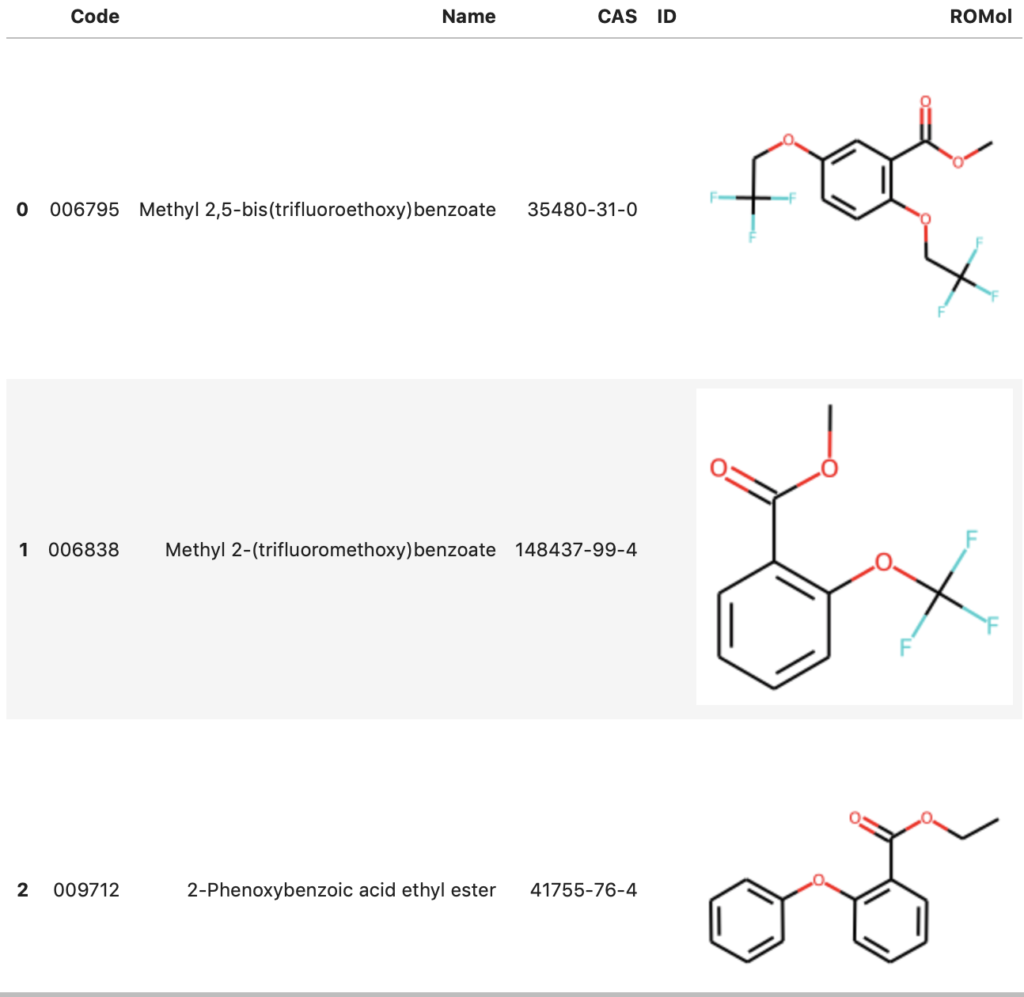
作成されたデータフレームには,Molオブジェクトが格納されているROMolカラムの他,カタログナンバーのCode,化合物名のName,化合物のCAS番号のCASなどのカラムがあることがわかります.
MOLオブジェクトへの水素原子の追加または除去
rdkit.Chem.RemoveHs(mol)
通常SMILESでは水素原子に関する情報は省略しますし,上述のようにSDFからの読み込みでは標準設定では水素原子なしでMolオブジェクトを作成します.
ここで紹介する2つのメソッドはMolオブジェクトに
- 水素原子を追加
- 逆に水素原子を除去
するのに使います.両方のメソッドともに,元のオブジェクトはいじらずに新しいMolオブジェクトを返します.
mol_benH = Chem.AddHs(mol_ben) print(Chem.MolToMolBlock(mol_benH, includeStereo=False))
RDKit
12 12 0 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
・・省略・・
M END
MOLブロックに水素原子の項が追加されているのが見てとれます.また「includeStereo=False」を設定しているため,原子の座標は「0」のままです.
2次元座標の計算
さきほどから扱っているベンゼンのMolオブジェクトには原子の座標が設定されていませんでした.そこで構造式描画用に2次元座標を計算してみましょう.
3次元構造については「RDKitによる3次元構造の生成」という記事を参照してください.
Compute2DCoordメソッドは指定したMolオブジェクトに座標をそのまま埋め込みます.下の例にあるように,includeStereo=Falseを指定しても,炭素のx,y座標が設定されていることが確認できます(z軸は0のままです).
from rdkit.Chem import AllChem AllChem.Compute2DCoords(mol_ben) print(Chem.MolToMolBlock(mol_ben, includeStereo=False))
RDKit 2D
6 6 0 0 0 0 0 0 0 0999 V2000
1.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7500 -1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.7500 -1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
・・省略・・
M END
終わりに
今回は「RDKitでケモインフォマティクスに入門」という内容について,
- ケモインフォマティクスとはどんな学問領域か?
- RDKitとはどんなソフトウェア・ライブラリか?
からはじめて,RDKitの基本的な使い方を説明してきました.具体的には,
- SMILESやMOLファイルをはじめとした,さまざまな入力形式からのMolオブジェクトの作成方法
- MOLオブジェクトに対する水素原子の付加や2次元座標の計算方法などの簡単な扱い方
- MolオブジェクトからSMILESやMolブロック形式への書き出し
について見てきました.MolオブジェクトはRDKitの中心をなす重要な要素です.次回は分子を構成する「原子」,「結合」,「環構造」などに着目しながらMolオブジェクトについてさらに理解を深めていきたいと思います.
>>次の記事:「RDKitの分子Molオブジェクトを扱う」
コメント