「RDKitの分子Molオブジェクトを扱う」という記事では,RDKitの分子であるMolオブジェクトを用いて2次元構造の取得と描画方法について学んできました.今回はそれを3次元構造へと展開していきます.
ほぼ全ての分子は立体的な性質を持ちますから,分子を3次元で考えることは大切です.
必要なライブラリのインポート
まずはいつものように必要なライブラリーのimportをしておきましょう.
from rdkit import rdBase, Chem from rdkit.Chem import AllChem, Draw from rdkit.Chem.Draw import IPythonConsole import sys, py3Dmol import pandas as pd print(sys.version) # 3.7.9 (default, Aug 31 2020, 12:42:55) # [GCC 7.3.0] print(rdBase.rdkitVersion) # 2020.09.1
3次元構造の生成
Molオブジェクトの2次元構造はAllChem.Compute2DCoodinateで計算することができました.それでは3次元構造はというと,AllChem.EmbedMoleculeで行うことが可能です.この際,RDKitには以下に示す4つの方法(と2つの改良法)が実装されています.2018.09からデフォルトの方法がETKDGに変更になっていることに注意してください.
| 方法 | コメント |
|---|---|
| ディスタンス・ジオメトリー(DG)法 | 2018.03までのデフォルト方法 |
| AllChem.KDG | useBasicKnowledge=True |
| AllChem.ETDG | useExpTorsionAnglePrefs=True |
| AllChem.ETKDG | useBasicKnowledge=True useExpTorsionAnglePrefs=True 2018.09以降のデフォルト方法 |
| AllChem.ETKDGv2 | 新しいパラメータを用いたETKDGメソッド.python3.6以降で利用可能 |
| AllChem.ETKDGv3 | マクロサイクルなど環状化合物の精度向上が計られたETKDGメソッド.2020.03以降で利用可能 |
立体配座生成の各アルゴリズムの説明
名前から想像できるように上記の3つは全てディスタンス・ジオメトリー(DG)法に改良を加えたものです.RDKitに実装されているDG法はStochasticな立体構造の生成方法としてはかなり精度がよいことがベンチマークによって示されていますが,芳香環が歪む傾向にあるなどといった明らかな難点も知られています.
そこでDG法で得た構造に対して,
- ベンゼン環は平面
- アルキンは直線
といった一般的な化学常識(basic knowledge)を加えたのがKDGになります.
またケンブリッジ結晶構造データベースの実験データから得た二面角の分布傾向(experimental torsion angle preferences)を考慮したものがETDGになります.そしてその両方を加えたのがETKDGです.
我々が普段使う分にはETKDGを使えばよいと思います.ETKDGについてさらに詳しく知りたい方は以下の論文を参照してください.
ディスタンス・ジオメトリー法で生成された分子は多くの場合,先に述べたように芳香環が歪んでいるなどあまり精度が高くよくありません.従ってDG法で得た構造は実際に分析を始める前に分子力学法(MM)で構造最適化する必要があります.分子力場としてはUFFとMMFFの2つがRDKitでは提供されています.
ETKDGでも構造の補正を行いますが,DG法による構造生成とMMを用いた最適化にかかる時間を考慮すると計算コストはそこまで変わらないとも言われています.
立体配座生成の各アルゴリズムの精度比較
今回は「Platinum dataset」と呼ばれるハンブルク大の研究チームが高精度のX線構造から得た構造データセットを用いて,
- DG法で発生させた構造をUFFで最適化した構造
- ETDG法で発生させた構造
- ETKDGで発生させた構造
- ETKDGv2で発生させた構造
- ETKDGv3で発生させた構造
の5種類について,もとのX線構造と比較した精度の違いを調べてみます.リンク先ウェブサイトの「Platinum Dataset 2017_01」を利用します.
なおPlatinum datasetに関しての詳細は以下の論文を参照してください.
「Benchmarking Commercial Conformer Ensemble Generators」(J. Chem. Inf. Model. 57, 11, 2719-2728.)
# 分子の読み込み
suppl = Chem.SDMolSupplier('./platinum_dataset_2017_01.sdf', removeHs=False)
mols = [x for x in suppl if x is not None]
len(mols) # 4548
# 分子を1度SMILESに変換して再構成
smiles = [Chem.MolToSmiles(mol) for mol in mols]
mols_from_sm = [Chem.MolFromSmiles(sm) for sm in smiles]
# それぞれの方法について4548分子の3次元構造を発生
def DG(mols):
DG_uff = []
mhs = [Chem.AddHs(mol) for mol in mols]
for mol in mhs:
AllChem.EmbedMolecule(mol,
useBasicKnowledge=False,
useExpTorsionAnglePrefs=False)
if AllChem.UFFHasAllMoleculeParams(mol):
AllChem.UFFOptimizeMolecule(mol)
DG_uff.append(mol)
return DG_uff
def ETDG(mols):
ETDG_mols = []
mhs = [Chem.AddHs(mol) for mol in mols]
for mol in mhs:
AllChem.EmbedMolecule(mol, AllChem.ETDG())
ETDG_mols.append(mol)
return ETDG_mols
def ETKDG(mols, version=1):
ETKDG_mols = []
mhs = [Chem.AddHs(mol) for mol in mols]
for mol in mhs:
if version == 1:
p = AllChem.ETKDG()
elif version == 2:
p = AllChem.ETKDGv2()
elif version == 3:
p = AllChem.ETKDGv3()
else:
print('invalid input')
AllChem.EmbedMolecule(mol, p)
ETKDG_mols.append(mol)
return ETKDG_mols
DG_m = DG(mols_from_sm)
ETDG_m = ETDG(mols_from_sm)
ETKDGv1_m = ETKDG(mols_from_sm)
ETKDGv2_m = ETKDG(mols_from_sm, 2)
ETKDGv3_m = ETKDG(mols_from_sm, 3)
# 結晶構造と発生させた構造の重原子RMDSを取得
RMSD = []
for i, (mol, dg, etdg, etkdg1, etkdg2, etkdg3) in enumerate(zip(mols, DG_m, ETDG_m, ETKDGv1_m, ETKDGv2_m, ETKDGv3_m)):
if i % 500 == 0:
print(i, 'th molecule')
mol = Chem.RemoveHs(mol)
dg = Chem.RemoveHs(dg)
etdg = Chem.RemoveHs(etdg)
etkdg1 = Chem.RemoveHs(etkdg1)
etkdg2 = Chem.RemoveHs(etkdg2)
etkdg3 = Chem.RemoveHs(etkdg3)
DG_rms = AllChem.GetBestRMS(mol, dg)
ETDG_rms = AllChem.GetBestRMS(mol, etdg)
ETKDG1_rms = AllChem.GetBestRMS(mol, etkdg1)
ETKDG2_rms = AllChem.GetBestRMS(mol, etkdg2)
ETKDG3_rms = AllChem.GetBestRMS(mol, etkdg3)
RMSD.append((DG_rms, ETDG_rms, ETKDG1_rms, ETKDG2_rms, ETKDG3_rms))
RMSD = pd.DataFrame(RMSD,
columns=['DG+UFF', 'ETDG', 'ETKDG', 'ETKDGv2', 'ETDKGv3'])
RMSD.describe().round(2)
| DG + UFF | ETDG | ETKDG | ETKDGv2 | ETKDGv3 | |
|---|---|---|---|---|---|
| count | 4548.00 | 4548.00 | 4548.00 | 4548.00 | 4548.00 |
| mean | 1.70 | 1.70 | 1.65 | 1.65 | 1.65 |
| std | 0.83 | 0.79 | 0.84 | 0.83 | 0.84 |
| min | 0.04 | 0.13 | 0.04 | 0.04 | 0.04 |
| 25% | 1.11 | 1.11 | 1.05 | 1.05 | 1.04 |
| 50% | 1.66 | 1.61 | 1.59 | 1.58 | 1.58 |
| 75% | 2.23 | 2.20 | 2.18 | 2.18 | 2.18 |
| max | 5.77 | 5.80 | 5.41 | 5.61 | 5.42 |
上記の結果からRMDSの平均値はETKDGシリーズが小さいことがわかります.DG+UFFやETDGと比べるとETKTGは精度がよい方法と述べてよさそうです.
IPython上で%timeを用いて手許の計算機で実行時間(CPU times)を計測した結果が以下の表になります.精度が高いだけでなく,DG+UFFと比べてETKDGシリーズの実行速度はやや速いと考えてよそうです.
| method | user | sys | total |
|---|---|---|---|
| DG + UFF | 3min 1s | 15.5 ms | 3min 1s |
| ETDG | 2min 10s | 28 ms | 2min 10s |
| ETKDG | 2min 42s | 12 ms | 2min 42s |
| ETKDGv2 | 2min 44s | 52 ms | 2min 44s |
| ETKDGv3 | 2min 51s | 20 ms | 2min 51s |
py3Dmolを用いた3次元構造の描画
続いて3次元構造を描画してみましょう.以前はPyMolを用いて描画するのが一般的だったと思いますが,py3Dmolの登場以降はJupyterノートブック上でこちらのライブラリを用いて描画する方が人気があるようです.
py3DMolをまだインストールしていない場合はcondaまたはpipでインストールしておきましょう.
conda install py3dmol
pip install py3Dmol

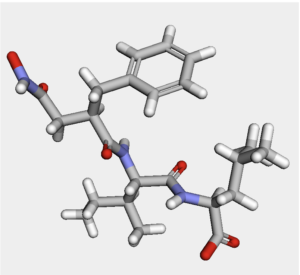
分子1つの描画
1つの分子を描画するにはdrawMol3Dを使うのが簡単です.描画したい分子のみを指定した場合はpy3DmolのViewが作成されて,そこに分子が表示されます.
IPythonConsole.drawMol3D(mols[0])
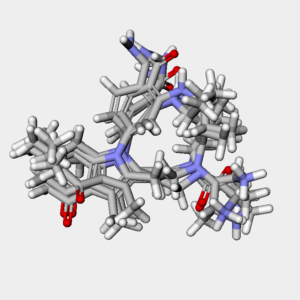
複数分子を1つの枠に描画
下のコードはpy3Dmolのviewオブジェクトを1つ作成して,そこに描画する分子を追加しています.その際にviewオブジェクトにはMolBlock形式かPDB形式で分子を渡す必要があるため,一度MolBlock形式に変換しています.IPythonConsoleを用いるとこの作業を内部でやってくれるわけです.
v = py3Dmol.view(width=400, height=400)
mols_show = [DG_m[30], ETDG_m[30], ETKDGv1_m[30], ETKDGv2_m[30]]
for mol in mols_show:
mb = Chem.MolToMolBlock(mol)
v.addModel(mb, 'sdf')
v.setBackgroundColor('0xeeeeee')
v.setStyle({'stick': {}})
v.zoomTo()
v.show()
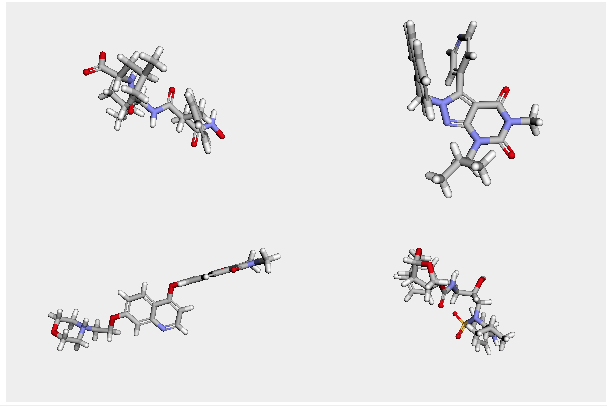
複数分子を違う枠に分けて描画
今度ののコードではviewオブジェクトをグリッドを用いて分割し,そこに1分子ずつ描画しています.linked=Falseオプションを使うことで,グリッド毎に分子を回転させることができるようになります.今回は静止画だけしかお示ししていませんが,是非自分の手で表示させてグリグリと回転させてみてみてください.
v = py3Dmol.view(width=600, height=400, linked=False, viewergrid=(2,2))
for m, i in zip(mols[:4], [(0,0),(0,1),(1,0),(1,1)]):
mb = Chem.MolToMolBlock(m)
v.addModel(mb, 'sdf', viewer=i)
v.setBackgroundColor('0xeeeeee')
v.setStyle({'stick': {}})
v.zoomTo()
v.show()
終わりに
今回はRDKitに実装されている3次元構造生成のアルゴリズムとpy3Dmolを用いた立体構造の描画方法について学びました.今回はどれだけ結晶構造に近い構造を得られるかに着目しましたが,立体構造の生成では分子が取り得る多様なコンフォマーを生成することも大事になります.
次回は1つの分子に対して複数のコンフォマーを発生させて,その構造分布を見ていきたいと思います.その過程でコンフォマーオブジェクトについて理解するとともに,今回簡単にだけ触れたRDKitにおける分子力学法の実装についても触れたいと思います.
>>次の記事:「RDKitによるコンフォマーの生成」
コメント
立体配座生成の各アルゴリズムの精度比較のセクションで
分子を一度SMILESに戻して再構成しているのには、どのような意図があるのですか?
コメントありがとうございます.
平面構造から立体構造を取得するという一般的用途を想定して,一度SMILESへと戻しています.
また生成される構造が初期構造に依存するかはわかりませんが,平面構造から発生させることで全ての手法を同一条件で比較しています.