
本サイトでは「有機合成化学者のためのケモインフォマティクス入門」を掲げて,高価なソフトウェアを用意することなく学習がはじめられるようにプログラミング言語pythonとそのケモインフォマティクス用ライブラリーであるRDKitを用いた解説をおこなっています.
「RDKitでフィンガープリントを使った分子類似性の判定」という記事では分子のフィンガープリントを導入することで分子の類似度を評価しました.類似度が高い分子の組み合わせは,化合物空間(ケミカルスペース)における距離が近いと言い換えることができます.この場合,「類似度」と「距離」は反対の意味に使われます.
今回の記事では化合物間の「距離」に着目し,化合物ライブラリーの中から,どのように似ていない分子を選び出すかについて学んでいきます.多様なライブラリーの構築は,ケモインフォマティクスにおける重要な課題ですので,さまざまな手法が考案されています.
化合物の類似性と多様性
分子類似性を議論した際に,「類似した化合物は似た性質を持つ」というsimilar property principleという考え方を紹介しました.ある生理活性を持つ化合物と似た構造の化合物はやはり同じような生理活性を持つというわけです.
すなわち,単一のライブラリーで広範な生理活性・性質をカバーしたい場合には,できるだけ「似ていない」化合物を集めることで達成できると考えられます.
問題は類似性の議論でも述べたように,何をもって「似ている」とするかは場合によって異なるので,万能な方法がないということになります.
化合物間の「距離」
化合物の類似性を評価する際に最も使われているのがタニモト係数で,その定義は以下の通りでした.
$$ S_{AB} = \frac{c}{a + b – c} $$
aは分子Aのビット配列で1が立っている数,bは分子Bのビット配列で1が立っている数,cは分子AとBで共通に1が立っている数を示します.タニモト係数はその定義から0から1の値を取ることになります.
化合物間の「類似度」とケミカルスペースにおける「距離」は反対の関係にありますので,
$$ D_{AB} = 1 – Tanimoto_{AB} $$
を定義すれば化合物間の距離も0から1の間を取ります.実際この値はSoergel距離と呼ばれ,タニモト係数とは独立して発表されました.
ライブラリー内の化合物ペアの距離が計算できるようになったので,どのように多様な化合物を選んでいくのがよいのか考えていきましょう.
総当たり方式による抽出は現実的に不可能
ライブラリー内の全ての化合物ペアの距離が得られるなら,「考えられる組み合わせをすべて考慮した上で,最も多様な化合物たちを選び出せばよい」と考える人もいらっしゃるかもしれません.しかし,このような総当たり方式(brute-force)のアプローチはすぐにうまくいかなくなります.
一般にN個の化合物を含むライブラリーからn個の化合物を選ぶ場合に,考えられる組み合わせは
$$ \frac{N!}{n!(N-n)!} $$
にも及びます.
これはたった50個からなるライブラリーから10個の化合物を選ぶだけで1010以上の組み合わせが存在することを意味します.
我々が興味があるライブラリーは通常50個よりもずっと大きなものですので,このような総当たり作戦は使い物にならないことが理解して頂けると思います.
幅広い化合物を抽出するための4つの方法
このようにありえる化合物の選び方は,あっという間に天文学的数字になるため,化合物を選ぶにあたって何らかの工夫が必要になります.この記事では
- クラスタリング法
- 距離ベースの方法
- 区分けをベースとした方法
- 最適化手法を用いる方法
といった4つの方法について述べていきます.
クラスタリング法
クラスタリングとは集団をいくつかの類似した小集団にわけることです.同じ集団に分けられた化合物は類似しているため,違う集団から化合物を選ぶことによって多様な化合物を選択することができます.
すなわちクラスタリング法を用いてライブラリー構築を行う場合は,
- 化合物をいくつかの集団にわける
- 各集団から化合物を選択する
という2段階を経ることになります.
クラスタリングの方法は分け方によって,
- 非階層型クラスタリング(Non-Hierarchical Clustering)
- 階層型クラスタリング(Hierarchical Clustering)
の2種類にわけられます.
非階層型クラスタリング
非階層型クラスタリングではデータセットをあらかじめ定めた数のクラスターに分割していきます.分割方法の異なるいくつかのアルゴリズムが知られています.
例えば下の図ではデータポイントを3つのクラスターへと分けています.この場合は3つに分割するのが最適とすぐに判断できますが,通常扱うような高次元のデータでは最適なクラスターの数は自明ではありません.

機械学習用ライブラリのscikit-learnにいつかの手法が実装されています.最も代表的な非階層型クラスタリング法であるk平均法を用いたクラスタリングについては「RDKitとk平均法による化合物の非階層型クラスタリング」という記事で解説しています.

階層型クラスタリング
階層型クラスタリングでもデータセットをいくつかのクラスターに分割していきますが,分割していく過程において樹形図のような階層構造があらわれます.
非階層型クラスタリングと同様に機械学習用ライブラリのscikit-learnにいくつかの手法が実装されています.最も代表的な凝集型クラスタリング法であるウォード法については「RDKitとウォード法による化合物の階層型クラスタリング」という記事で解説しています.

そこでこの記事ではRDKitに実装されているウォード法の使い方をみていきたいと思います.
RDKitにおけるウォード法の実装
rdSimDivPickers.ClusterMethod
RDKitでは多様なライブラリー構築のための手法はSimDivFilters.rdSimDivPickersに,クラスタリング法に関するコードはML.Clusterに実装されています.今回は前者にあるHierarchicalClusterPickerを使ってウォード法で化合物を選択してみます.
分子の準備
分子は以前のscikit-learnの場合と同様に,今回もナミキ商事の運営するChemCupidからダウンロードした,カタログの新規追加分の分子を収めたSDFを使って,ランダムにシャッフルした5000個を使います.
### 必要なライブラリのインポート
from rdkit import rdBase, Chem, DataStructs
from rdkit.Chem import AllChem
import numpy as np
### 分子の読み込み
suppl = Chem.SDMolSupplier('./Namiki_NEW_201807_0001.sdf')
mols = [x for x in suppl if x is not None if x.GetProp('NScode').endswith('0000')]
len(mols) ### 66005
### フィンガープリントの作成
morgan_fps = [AllChem.GetMorganFingerprintAsBitVect(m, 2, 2048) for m in mols]
ウォード法によるクラスタリング法の実行
HierarchicalClusterPickerのPickメソッドは
- numpy配列型の距離行列
- もとの集団の大きさ
- 選ぶ化合物の大きさ
の3つを引数としてとります.注意すべき点は距離行列としては,全行列のうち下半分を1つのリストにしたものを与えます.つまり,下の図の灰色で囲んである部分を赤い数字の順番に1つのリストにします.ここを間違えると結果が変わってきてしまいますので注意してください.

HierarchicalClusterPickerではインスタンスの生成段階でどの方法でクラスタリングを行うかを設定できます.下のコードではClusterMethod.WARDによってウォード法を指定します.
from rdkit.SimDivFilters.rdSimDivPickers import HierarchicalClusterPicker, ClusterMethod
### 距離行列の計算
dis_mat = []
for i in range(1,5000):
dis_mat.extend(DataStructs.BulkTanimotoSimilarity(morgan_fps[i], morgan_fps[:i], returnDistance=True))
### HierarchicalClusterPickerインスタンスの生成
ward = HierarchicalClusterPicker(ClusterMethod.WARD)
ids = ward.Pick(np.array(dis_mat), 5000, 6)
list(ids)
Pickでは化合物IDがリストでもどってきます.RDKitを使うと形式的には1段階ですが,内部では「クラスタリング」と「分子の選択」という2段階プロセスを経ています.
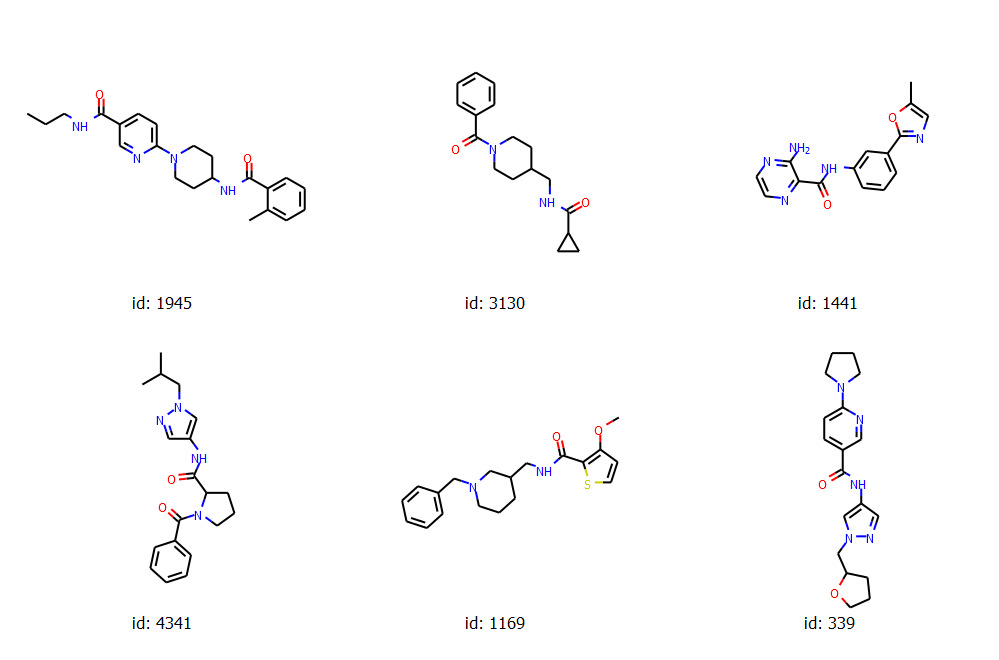
[1945, 3130, 1441, 4341, 1169, 339]
選ばれた化合物の構造はこちらのようになりました.
距離ベースの方法
距離ベースの方法はクラスタリング法とは異なり,1段階で分子の選択をすませる方法になります.その選択プロセスは
- データセットから分子を選び,小集団に入れる
- データセット中の分子同士,および小集団中の分子の距離を計算する
- 次の化合物を,小集団中の分子と「最も遠い」分子を選択する
- 小集団の分子数が指定数に達していないならば,2に戻る
という段階を繰り返すことで行います.色々なアルゴリズムが存在しますが,
- 最初にどのように分子を選ぶか
- どのように「最も遠い」分子を決めるか
の2点がポイントです.「遠い分子」の選定には特にMaxSum法とMaxMin法という2つの方法がよく使われます.
MaxSum法
MaxSum法では小集団中にm個の分子が存在する際に
$$ score_{i} = \sum_{j=1}^{m} D_{i,j} $$
という関数を最大化する分子を選択します.ここでDi,jは2つの分子iとjの間の距離です.つまり小集団中の全ての分子との距離の和を考えて次の分子を選んでいきます.
MaxMin法
MaxMin法では,同様に小集団中にm個の分子が存在する際に
$$ score_{i} = minimum (D_{i,j: j=1,m}) $$
を最大化する分子を選択します.つまり,小集団中の分子から最も離れている分子を選んでいきます.MaxMin法では小集団内のうち最近接の分子のみに注目します.
「RDKitのMaxMin法を用いた多様性の高いライブラリの選定」という記事では,RDKitに実装されているMaxMin法について解説しています.
区分けをベースとした方法
クラスタリング法と距離ベースの方法はどちらも,「個々の分子間の距離(または類似度)」に注目した方法になります.そのため,当然ですが用いるデータセットによって分類法が異なります.
区分けをベースとした方法では,事前に決めた基準に沿って分子を分類する方法になります.通常この基準は2つか3つ程度の少ない特徴量を区分けしたもので,そのため空間内をセルで区切ったように可視化することが可能になります.
利点としては,
- データセット中で疎な部分を容易に認識できる
- 異なるデータセット間の違いを,重なり具合を比較することで判断できる
といった点があげられます.どちらもクラスタリングや距離ベースの方法では,すぐに得ることの難しい点になります.
反対に欠点としては,
- 基準とできる特徴量が少ないため,低次元での表現に限られる
- セルの区切りが恣意的であり,小さな変更で結果が大きく変わることがある
といった点があげられます.基準とする特徴量が限られているため,本当に目的に沿ったものにする必要があります.
ZINC15の例
この分類法の例としては,バーチャルスクリーニング用に市販化合物を集めたZINC15データベースがあげられます.下のように化合物を「logP」と「分子量」という2つの基準に分けて,化合物を切り分けています.
本データベースの主な利用目的は「バーチャルスクリーニング」ですので,物性・活性に関係し,かつ計算も容易な「logP」と「分子量」を基準とした区分けは有効だと考えられます..

化合物の数を見ることで「疎な部分」と「密な部分」がわかりますが,ここではさらにヒートマップを活用することで視認性を向上させています.とてもうまい可視化方法だと思います.
最適化手法を用いる方法
「ある集団から一定個数を,その多様性が最大になるように選ぶ問題」は,選んだ化合物間の距離に関連した関数(例えば距離の和)の最適化問題と見なすことができます.
このような考え方に基づいてモンテカルロ法にシミュレーティド・アニーリングなどを組み合わせて最適化問題を解いていくことが行われています.このアプローチでは,ランダムに選んだ集団からはじめて,集団内の複数分子を同時に組み替えながら,効率的に多様性を最大化していくことが期待できます.
多様性を表現する関数型としては上で述べたMaxSumやMaxMinが多いですが,新しい関数型の開発による効率性を追求も重要な研究テーマになっています.
終わりに
今回は「ケモインフォマティクスで多様な化合物ライブラリーを構築する」という話題について解説してきました.
- 分子の「類似性」とケミカルスペースにおける「距離」の関係
- どのように多様性の高い化合物ライブラリーを構築するかに関する4つの方法
について扱いました.特によく使われるクラスタリング法とMaxMin法については具体例を使って理解を深めておくのがよいと思います.
これまでRDKitの基本的な使い方の説明からはじめて,ケモインフォマティクスらしい分析を行ってきました.次回はまた化合物の扱い方に戻り,RDKitでどのように化学反応を扱うかを見ていきたいと思います.
>>次の記事:「RDKitで化学反応:ケモインフォマティクスにおける反応式の扱い方」
コメント
HierarchicalClusterPickerのPickメソッドで得られる化合物IDのリストを、構造式として表示する手法について質問です。この化合物IDは元々のmolsの化合物番号と対応しているのでしょうか?例えば、下記のようなコードで良いのでしょうか?よろしくお願いいたします。
Draw.MolsToGridImage([mols[i] for i in [1945, 3130, 1441, 4341, 1169, 339]])
ご質問ありがとうございます.
> この化合物IDは元々のmolsの化合物番号と対応しているのでしょうか?
対応していると考えてよいです.本文の構造はまさにご提示頂いた方法で描画しています.
今後ともよろしくお願いいたします.
管理人