
当サイトではこれまで多数の記事でケモインフォマティクス用のライブラリであるRDKitの使い方を説明してきました.これまで扱った内容は化合物の表現方法,構造,性質をはじめとして多岐にわたりますが,主に化合物1つ1つを,時には集団として,どのように扱うかに焦点を当ててきました.
今回は化合物間の変換,すなわちケモインフォマティクスで化学反応をどのように取り扱うかについて学んでいきます.
まずは以前に取り扱ったMOLファイルやSMILES記法といった単一の化合物の表記法を,化学反応を表現するためにどのように拡張していくかを学びます.続いてRDKitでの化学反応の扱い方を学習していきましょう.
MOLファイルやSMILES記法について不安がある方は以下の記事を先に読むことをおすすめします.


RXNファイル
「MOLファイル・SDFとはどんな化学情報ファイルなのか?」という記事では「MOLファイル」,及びそれを拡張して複数分子を取り扱い可能にした「SDF」について説明しました.
MOLファイルのフォーマットを用いて化学反応を表現するように拡張したものが,RXNファイルと呼ばれる形式です.基本構成単位は同じですので,MOLファイルやSDFを取り扱い可能なソフトウェアならRXNファイルも扱えるのが通常です.
例えば次のような酢酸とメチルアミンのアミド化反応を考えてみます.

ChemDrawでスケッチしたこのファイルを「save as」で保存しようとすると,下のように「Reaction Molfile (RXN)」が選択可能です.

保存したRXNファイルの中身は以下のようになっています.空行が多いですが,構成としては
- 「$RXN」からはじまるヘッダー部分
- 「$MOL — $END」から成る分子数分のブロック
という形になっています.ヘッダー部分の「2 1」という部分が原系(矢印の左側)と生成系(矢印の右側)の分子数を表しています.
$RXN
ChemDraw00000000000D
2 1
$MOL
4 3 0 0 0 0 0 0 0 0999 V2000
-0.7145 -0.6187 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0000 -0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7145 -0.6187 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.0000 0.6187 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0
2 3 1 0 0
2 4 2 0 0
M END
$MOL
2 1 0 0 0 0 0 0 0 0999 V2000
-0.4125 0.0000 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
0.4125 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0
M END
$MOL
5 4 0 0 0 0 0 0 0 0999 V2000
-1.0705 -0.6187 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3560 -0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3584 -0.6187 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.3560 0.6187 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
1.0705 -0.2076 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0
2 3 1 0 0
2 4 2 0 0
3 5 1 0 0
M END
例えば次のように生成系に水分子を加えた反応式をRXNファイルとして保存するとヘッダー部分が変わっているのが確認できます.

$RXN ChemDraw00000000000D 2 2 $MOL ・・・(以下略)・・・
Reaction SMILES/SMARTS
「SMILES記法は化学構造の線形表記法」という記事では,化合物を文字列で扱う方法の1つとして「SMIELS」というものを説明しました.このSMILES記法を化学反応を記述するために拡張したものが,「Reaction SMILES」になります.
Reaction SMILESでは主に
- 原料・生成物などの反応の構成要素の指定
- 反応する原子のナンバリングによる指定
という2種類の拡張がなされています.順番に見ていきましょう.
Reaction SMILESにおける反応の構成要素
原料・試薬と生成物を区別するためにReaction SMILESでは「>」を区切りとして用います.右矢印(→)の簡略化されたものと覚えるとよいでしょう.
一般的な表現形式としては下のような形になります.
この際,左辺と右辺で原子数が一致していなくても有効なSMILESとなります.即ち,例えばアミド化反応で生成する水分子や,鈴木–宮浦クロスカップリングで生じるホウ素含有の共生成物などはSMILESに含めなくてもよいということです.
また触媒・溶媒などを記載しない場合には
のように,「>>」を区切りとして用います.実際,この形式での表現を見かけることが一番多いですし,後ほど見るようにRDKitでもこの形式のSMARTSを用いて反応を扱います.
それでは具体例として,下記のような求核置換反応を考えてみましょう.

このSN2反応のReaction SMILESとしては例えば下の表のように表記することができます.何れの表記も正しいSMILESです.
| 説明 | Reaction SMILES |
|---|---|
| 原料と生成物のみ | BrCC=C>>C=CCC#N |
| 試薬と副生成物を追加 | [K+].[C#N-].BrCC=C>>C=CCC#N.[K+].[Br-] |
| 触媒・溶媒も追加 | [K+].[C#N-].BrCC=C>[Na+].[I-].[H]C(N(C)C)=O>C=CCC#N.[K+].[Br-] |
Reaction SMILESにおける原子のナンバリング
先ほどの求核置換反応では,SN2反応とSN2’反応という2つの反応機構が考えられます.このようにどの原子が反応するかを明示的に指定したい場合には,原子のナンバリングが大切になります.
原子のナンバリングには[元素記号+水素・電荷などの修飾情報:番号]のようにブラケット([])で囲んだ最後に「:1」のように指定します.
上の例ではアリル位での置換反応なら,
- Br[C:1]C=C>>C=C[C:1]C#N
のように,逆にオレフィン炭素からのSN2’なら,
- Br[C:1][C:2]=[C:3]>>[C:1]=[C:2][C:3]#N
のように表記できます.
RDKitでの化学反応の取り扱い
これまでケモインフォマティクスでの化学反応の扱い方としてRXNファイルとReaction SMILESについて見てきました.RDKitでも,RXNファイルとSMILESを拡張したSMARTS記法に基づいたReaction SMARTSからReactionオブジェクトを組み立てることができます.
Reactionオブジェクトの生成
AllChem.ReactionFromSmarts(rxn-smarts)
AllChem.ReactionToRxnBlock(rxn-obj)
AllChem.ReactionToSmarts(rxn-obj)
先ほど述べたようにReactionオブジェクトは
- RXNファイル
- Reaction SMARTS
から作成可能で,またこれらの形式への出力が可能です.
下の例では先ほどみたアミド化反応のRXNファイル2種類を読み込み,GetNumReactantTemplatesとGetNumProductTemplatesメソッドを使って,原系と生成系の数を表示しています.
from rdkit import Chem
from rdkit.Chem import AllChem
rxn = AllChem.ReactionFromRxnFile('./amide_rxn.rxn')
rxn2 = AllChem.ReactionFromRxnFile('./amide_rxn2.rxn')
type(rxn)
rxn.GetNumReactantTemplates(), rxn.GetNumProductTemplates()
rxn2.GetNumReactantTemplates(), rxn2.GetNumProductTemplates()
rdkit.Chem.rdChemReactions.ChemicalReaction (2, 1) # for rxn (2, 2) # for rxn2
Reactionオブジェクトで反応の実行
作成したReactionオブジェクトを用いて反応を行うにはRunReactantsメソッドを使用します.またMolオブジェクトが反応基質となるか否かのチェックにはIsMoleculeReactantメソッドで調べることが可能です.
下の例では酢酸とメチルアミンではなく,カルボン酸と脂肪族アミンとの縮合反応をSMARTS記法を用いて記述し,Reactionオブジェクトを作成しています.
benzoic_acid = Chem.MolFromSmiles('c1ccccc1C(=O)O')
benzylamine = Chem.MolFromSmiles('c1ccccc1CN')
smarts = '[C:1](=[O:2])O.[N!0H:3][C:4]>>[C:1](=[O:2])[N!0H:3][C:4]'
rxn3 = AllChem.ReactionFromSmarts(smarts)
x = rxn3.RunReactants([benzoic_acid, benzylamine])
len(x) ### 1
p = x[0][0]
反応の結果は考えられる生成物を網羅したリストになっていて,各リストにリストが内包されていてMolオブジェクトが格納されています(x[0][0]の部分).今回は反応点は各々1つしかありませんので,リストxの長さは1になります.
得られる化合物は当然以下のとおりです.

具体例:鈴木・宮浦クロスカップリング
有機化学者が日常的に用いる化学反応と言えば,アミド化と鈴木・宮浦クロスカップリングが代表的です.ここでは後者を例にして,臭化アリールとボロン酸ピナコールエステルを含むSDFを使って,ランダムにいくつかの基質をカップリングさせてみましょう.
使用する分子
下のような構造検索によって東京化成から取得した次のSDFを用います.


suppl1 = Chem.SDMolSupplier('./sdf_ArBr.sdf')
ArBr = [x for x in suppl1 if x is not None]
len(ArBr) ### 100
suppl2 = Chem.SDMolSupplier('./sdf_Bpin.sdf')
Bpin = [x for x in suppl2 if x is not None]
len(Bpin) ### 190
分子数は臭化アリールが100,ボロン酸エステルが190でした.このうち,ポリマー生成を防ぐために以下の構造を有するものを除いておきましょう.
- 臭化アリールのうちヨウ素を含むもの
- 臭化アリールのうち臭素を3個以上含むもの
- ボロン酸エステルで臭素またはヨウ素を含むもの
- ボロン酸エステル部位を分子内に複数持つもの
コードとしては「RDKitの分子Molオブジェクトを扱う」という記事で学んだようにアトムオブジェクトのGetSymbolメソッドを使うことで原子の種類を判定しています.
def atomic_symbol_in_mol(symbol, mols):
### symbol: atomic symbol for check
### mols: list of molecules
for mol in mols:
atom_list = []
for atom in mol.GetAtoms():
atom_list.append(atom.GetSymbol())
if symbol == 'I':
if atom_list.count('Br') < 3: ### 臭素数でスクリーニング
if not 'I' in atom_list:
reactants_1.append(mol)
if symbol == 'B':
if atom_list.count('B') < 2: ### ボロン酸数でスクリーニング
if not ('Br' in atom_list or 'I' in atom_list):
reactants_2.append(mol)
### 基質構造によるスクリーニング
reactants_1 = []
reactants_2 = []
atomic_symbol_in_mol('I', ArBr)
atomic_symbol_in_mol('B', Bpin)
len(reactants_1), len(reactants_2) ### (86, 153)
鈴木・宮浦クロスカップリングの定義
続いて実施するカップリング反応を定義します.ボロン酸エステルとしては脂肪族のものも含まれていますが,芳香族ボロン酸のみを反応基質とすることにします.さらに基質をシャッフルした上で最初の20個ずつを反応に使用することとしましょう.
import numpy as np ### 反応の定義 suzuki_rxn = '[c:1][c:2](Br)[c:3].[c:4](B(O)O)>>[c:1][c:2]([c:4])[c:3]' rxn4 = AllChem.ReactionFromSmarts(suzuki_rxn) ### 基質のシャッフル np.random.seed(1234) np.random.shuffle(reactants_1) np.random.shuffle(reactants_2) sub1 = reactants_1[:20] sub2 = reactants_2[:20]
カップリング反応の実行
得られた基質たちを単純に二つのfor文で入れ込んで総当たりをすることでカップリング体が得られますが,今回はもう少し複雑にしてみることにします.
基質中に複数の臭素が存在する場合(今回は1つか2つ)に,生成物に対してさらにカップリング反応を行ってみましょう.そのためには,最初に生じた生成物が反応の基質になるかをIsMoleculeReactantメソッドでチェックします.
具体的には生成物が反応基質であった場合に,再帰的にカップリング関数を呼び出すことで実現可能です.その際に両基質で反応点が複数あるとポリマーが生成して無限ループに入り込んでしまうために,最初に基質構造でスクリーニングを行いました.
また違う基質を用いたカップリング反応から同じ生成物が得られる可能性を考えて重複を除く関数を定義しています.
def cross_coupling(mol1, mol2):
### mol1: ArBr, mol2: Bpin
product_smiles = {}
product_list = []
compounds = rxn4.RunReactants([mol1, mol2])
if len(compounds) == 0:
return product_list
for compound in compounds:
sm = Chem.MolToSmiles(compound[0])
product_smiles[sm] = compound[0]
### 重複化合物を取り除く操作
for smiles in product_smiles.keys():
p = Chem.MolFromSmiles(smiles)
product_list.append(p)
### 2回目のカップリングを行うか否かのチェック
if rxn4.IsMoleculeReactant(p):
p_01 = cross_coupling(p, mol2)
p_02 = cross_coupling(mol1, p)
if not p_01 == []:
product_list.extend(p_01)
if not p_02 == []:
product_list.extend(p_02)
return product_list
def remove_duplicates(mols):
smiles_dict = {}
final_mols = []
for mol in mols:
sm = Chem.MolToSmiles(mol)
smiles_dict[sm] = mol
for smiles in smiles_dict.keys():
m = Chem.MolFromSmiles(smiles)
AllChem.Compute2DCoords(m)
final_mols.append(m)
return final_mols
### カップリングの実行
products = []
for i in sub1:
for j in sub2:
products.extend(cross_coupling(i,j))
print(len(products)) ### 418
products = remove_duplicates(products)
print(len(products)) ### 418
全部で418種類のカップリング体が得られたようです.一部の構造はこちらに示します.
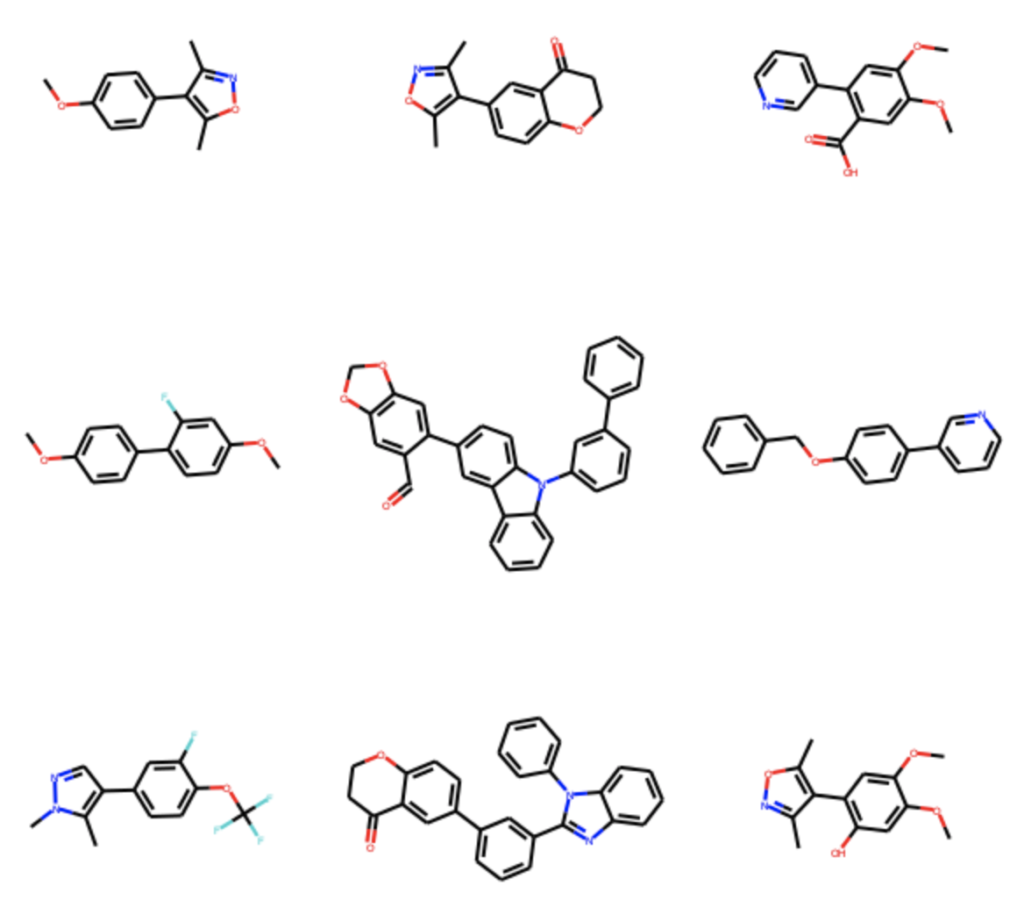
終わりに
今回はケモインフォマティクスにおいて化学反応をどのように表現するかの基本から,実際にRDKitを用いた取り扱い方を見てきました.最後に鈴木・宮浦クロスカップリングをランダムに行わせることで生成物のライブラリを作成してみました.
段々と複雑なことができるようになってきましたが,基本的にはこれまで学んだことを1つ1つ積み重ねているだけです.少し歩みが速すぎると感じた方は,「RDKitでケモインフォマティクスに入門」から復習してみるのもよいかと思います.
次回は部分構造・フラグメントに着目して化学反応を掘り下げていきたいと思います.
>>次の記事:「RDKitでRECAPを用いた分子のフラグメント化」
コメント