
「RDKitでフィンガープリントを使った分子類似性の判定」という記事では分子のフィンガープリントを導入することで,分子同士の類似度を評価しました.また原子ごとの寄与度を類似度マップを用いて可視化する方法を「RDKitの類似度マップを用いて原子ごとの寄与を可視化する」という記事で学びました.
今回は「似ている分子は共通構造を持つ」という考えに則り,部分構造検索から主鎖・側鎖への分離などの構造変換を扱います.さらに化合物の主鎖と側鎖に関連して,分子骨格(scaffold)というものを見ていきましょう.そして最後に最大共通部分構造(Maximum Common Substructure/Subgraph; MCS)という考え方へと展開させていきます.いずれも今後大量の化合物を扱っていく際に重要な考え方になります.
分子の準備
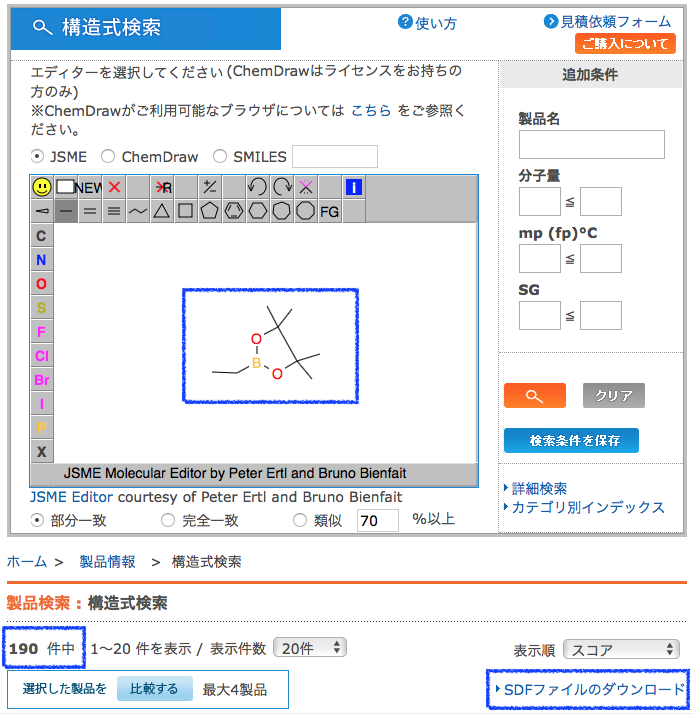
まずは必要なライブラリのimportと分子を用意しましょう.今回は東京化成で市販されているボロン酸ピナコールエステルのSDFを使って見ましょう.全部で190化合物がヒットしました.
from rdkit import rdBase, Chem
from rdkit.Chem import AllChem, Draw
from rdkit.Chem.Scaffolds import MurckoScaffold
print(rdBase.rdkitVersion) ### 2017.03.3
suppl = Chem.SDMolSupplier('./sdf_20180823084638.sdf')
mols = [x for x in suppl if x is not None]
len(mols) ### 190
部分構造検索
Mol.GetSubstructMatch(query)
Mol.GetSubstructMatches(query)
「RDKitの分子Molオブジェクトを扱う」という記事でも簡単に説明したように,RDKitのMolオブジェクトには部分構造検索を行うメソッドが実装されています.
2つのメソッドがありますが,注意点としてはどちらのメソッドも検索構造(query)はMolオブジェクトにする必要があるという点が挙げられます.部分構造ですが分子Molに変換してください.
部分構造マッチの有無
HasSubstructMatchでは,指定した部分構造を持つか否かが返ってきます.例えばこのメソッドを使うことで,化合物の簡単なスクリーニングを行うといった利用が考えられます.
下の例ではSDFからフェニルボロン酸ピナコールエステル部位を有する化合物だけを取りだしています.今回の場合は,へテロ環や脂肪族ボロン酸エステルを取り除くことで190から139化合物へと絞り込みました.
### テンプレートのPhBpinを準備
ph_bpin = pcp.get_compounds('phenyl boronic acid pinacol ester', 'name')
len(ph_bpin) # 1
ph_bpin = ph_bpin[0]
tmp = Chem.MolFromSmiles(ph_bpin.canonical_smiles)
### 部分構造マッチ
ph_mols = [x for x in suppl if x.HasSubstructMatch(tmp)]
len(ph_mols) # 139
マッチした部分構造の取得
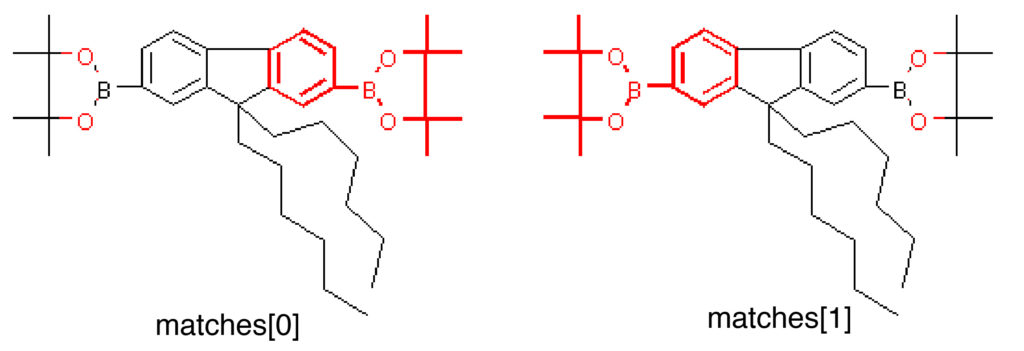
続いて具体的にマッチした構造を取り出してみましょう.GetSubstructMatchでは最初にマッチした部位の原子インデックスが,GetSubstructMatchesでは複数マッチした場合にも全ての原子インデックスがタプルとして取得できます.これをDraw.MolToImageのhighlightAtomsオプションへと渡すことで,マッチした部位を表示することが可能です.下の例では2つのピナコールエステル部位がヒットしています.
matches = ph_mols[4].GetSubstructMatches(tmp) Draw.MolToImage(ph_mols[4], highlightAtoms=matches[0]) Draw.MolToImage(ph_mols[4], highlightAtoms=matches[1])
((17, 13, 14, 15, 16, 23, 24, 25, 26, 10, 9, 7, 8, 12, 11), (22, 18, 19, 20, 21, 29, 30, 27, 28, 4, 3, 2, 1, 0, 5))
部分構造マッチを用いた構造変換
AllChem.ReplaceSubstructs(mol, query, replacement)
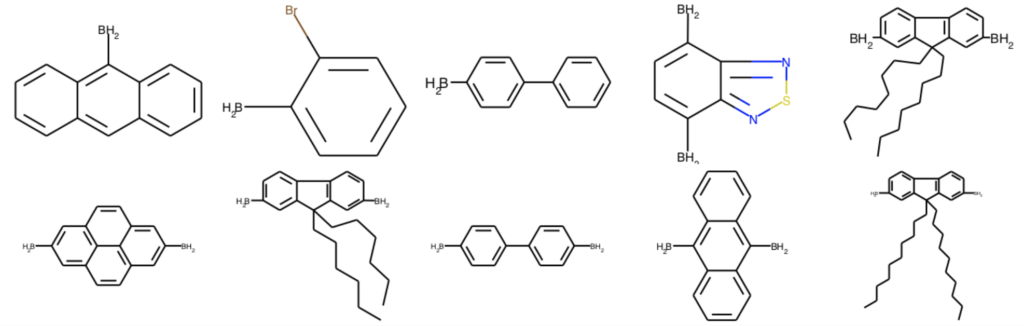
部分構造マッチを用いた構造変換では,削除(Delete)と置換(Replace)が可能です.何れのメソッドもオリジナルの分子には変更を加えずに,新しいMolオブジェクトを返します.ここではボロン酸エステルのピナコール部位を除去してみます.
delete_mols = [AllChem.DeleteSubstructs(x, Chem.MolFromSmiles('CC(C)(C(C)(C)O)O')) for x in ph_mols]
Draw.MolsToGridImage(delete_mols[:10], molsPerRow=5)
分子の主鎖と側鎖への分離
AllChem.ReplaceSidechains(mol, core)
Chem.GetMolFrags(mol ,asMols=False)
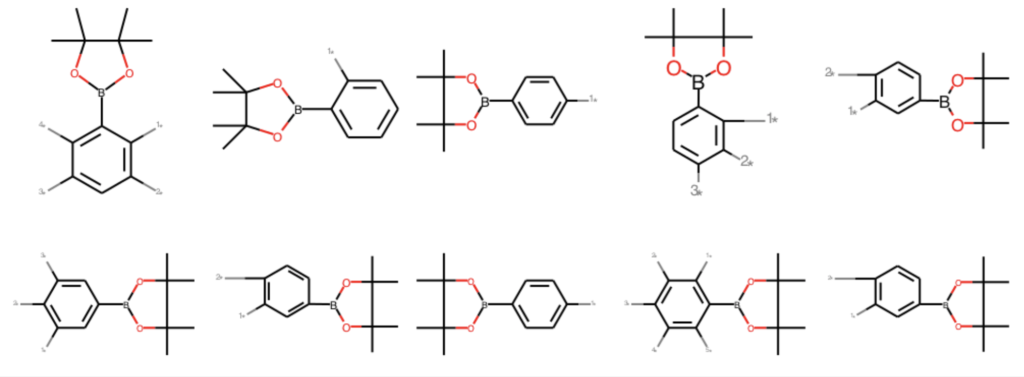
部分構造検索の際に,検索構造を主鎖(core),その他を側鎖(sidechains)と定義することで,構造の分離が可能になります.例えばフェニルボロン酸ピナコールエステルを主鎖として側鎖を除去し,ダミーアトムで置き換えたものが下の例になります.
replace_side = [AllChem.ReplaceSidechains(x, tmp) for x in ph_mols] Draw.MolsToGridImage(replace_side[:10], molsPerRow=5)
反対に主鎖を除去した場合は,置換様式によっては複数の側鎖が得られます.
replace_core = [AllChem.ReplaceCore(x, tmp) for x in ph_mols] Chem.MolToSmiles(replace_core[67]) ### '[*]N.[*][N+](=O)[O-]' Draw.MolsToGridImage([ph_mols[67], replace_side[67], replace_core[67]])
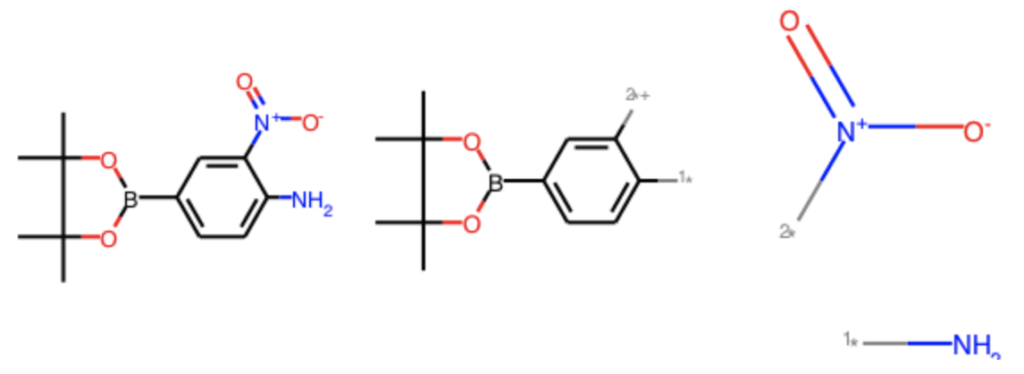
ReplaceCoreで得られた側鎖構造は1つのMolオブジェクトになっています.これを側鎖構造ごとに分割するにはGetMolFragsを使います.デフォルト設定では原子Idが返ってきますが,asMols=Trueオプションを使うことでMolオブジェクトとして得ることが可能になります.
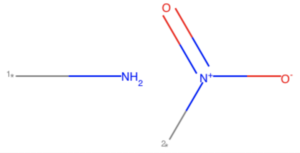
今回の場合はアミノ基とニトロ基に分けて得られます.
side_mol = Chem.GetMolFrags(replace_core[67], asMols=True) Draw.MolsToGridImage([x for x in side_mol])
RDKitでの分子骨格:Bemis/Murcko (BM) scaffolds
Scaffolds.MurckoScaffold.MakeScaffoldGeneric(mol)
先ほどの例で分子を主鎖と側鎖へと分離しました.RDKitにはBemis・Murckoらによる方法を使って分子の主骨格を取り出す方法が実装されています.例えば以下の4分子は我々の目には側鎖のの形・長さが違うだけに見えます.GetScaffoldForMolを使うことで,まさに我々が思ったとおりの骨格が取得可能です.
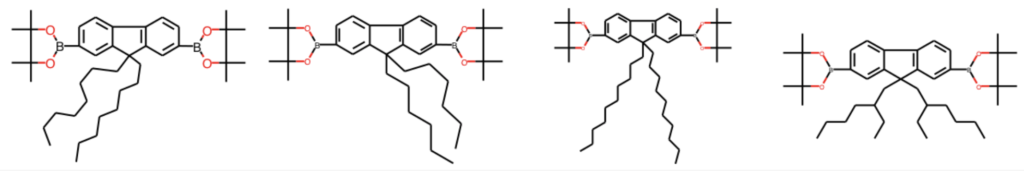
compounds = [4,6,9,14] scaffold1 = [MurckoScaffold.GetScaffoldForMol(ph_mols[i]) for i in compounds] Draw.MolsToGridImage(scaffold1, molsPerRow=4)

では次の2つの分子はどうでしょうか?我々の目にはオキサジアゾールとチアジアゾールはあまり変わらないですね.MakeScaffoldGenericはヘテロ原子を全て炭素に変えた骨格を返します.そのため,ベンゼンとピリジン,フランとピロールといった構造が似ているものを同一骨格として扱うことが可能になります.

Draw.MolsToGridImage([MurckoScaffold.MakeScaffoldGeneric(ph_mols[i]) for i in (23,30)])

MCS (Maximum Common Substructure/Subgraph),最大共通部分構造とは?
これまで見てきた部分構造検索では入力構造として検索したい構造を知っている必要があります.RDKitでは複数分子を入力構造として,共通部分構造(MCS)を探索するアルゴリズムが実装されています.
FindMCSでは様々なオプションによって,非常に細かく探索構造を制御することができます.ここではいくつかの要素について解説しますが,詳細は「Maximum Common Substructure」や「Module rdFMCS」などを参照してください.
| オプション | 意味 |
|---|---|
| atomCompare | |
| CompareAny | 原子の種類に関係なくマッチ |
| CompareElements | 原子の種類でマッチ(デフォルト) |
| CompareIsotopes | 同位体の種類でマッチ |
| bondCompare | |
| CompareAny | 結合の種類に関係なくマッチ |
| CompareOrder | 同じ結合でマッチ.ただし,芳香族と単結合は互いにマッチ(デフォルト). |
| CompareOrderExact | 同じ結合でのみマッチ |
| ringMatchesRingOnly | |
| False | デフォルト |
| True | 環内の結合は環内の結合のみとマッチ |
| completeRingsOnly | |
| False | デフォルト |
| True | 環構造が一致する場合のみマッチ |
2つの分子構造(グラフ)から最大の共通構造(グラフ)を探す問題はNP-complete問題に属するため,一般にMCSを探すには長い計算時間を必要とします.
そのためFindMCSメソッドではtimeout(=3600)オプションを用いることで,どの程度時間が経った場合には探索を終了するかを指定可能です(cpu timeではなく,wall-timeでの設定になります).
それではいくつか例を見ていきましょう.
MCSResultオブジェクト
MCSResult.numBonds
MCSResult.smartsString
FindMCSメソッドはMCSResultオブジェクトを返します.このオブジェクトからは原子数と結合数を取得したり,取得構造のSmartsを取り出すことが可能です.
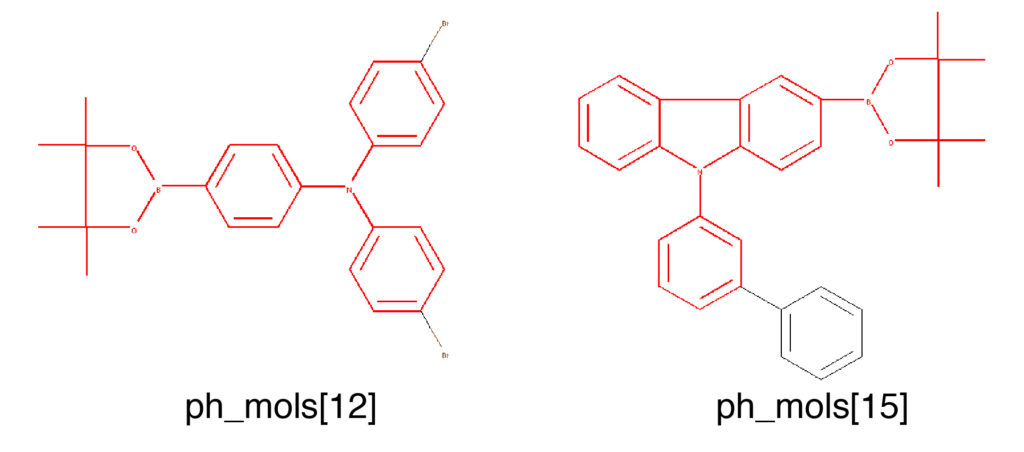
下の例では標準設定でMCSを探索した後に,SmartsからMolオブジェクトを作成し,部分構造マッチを用いて構造をハイライトしています.
mcs_01 = rdFMCS.FindMCS([ph_mols[12], ph_mols[15]]) mol_mcs_01 = Chem.MolFromSmarts(mcs_01.smartsString) Draw.MolToImage(ph_mols[12], highlightAtoms=ph_mols[12].GetSubstructMatch(mol_mcs_01)) Draw.MolToImage(ph_mols[15], highlightAtoms=ph_mols[15].GetSubstructMatch(mol_mcs_01))
MCSの構造探索オプション例
下の例ではホウ素のp位に位置する窒素周りの扱いが焦点になります.
デフォルト設定のMCSでは窒素及びそこから伸びる芳香環がマッチしています.一方でbondCompareをCompareOrderExactにした場合にはカルバゾール(分子A)とアニリン(分子B)では芳香族と単結合で結合次数が変わってきますのでマッチしてきません.
またringMatchesRingOnlyの場合も,カルバゾール窒素は環内の原子であるのに対し,アニリンは環を形成していませんのでマッチしてきません.
f1 = rdFMCS.FindMCS([ph_mols[39], ph_mols[44]]) f2 = rdFMCS.FindMCS([ph_mols[39], ph_mols[44]], bondCompare=rdFMCS.BondCompare.CompareOrderExact) f3 = rdFMCS.FindMCS([ph_mols[39], ph_mols[44]], ringMatchesRingOnly=True) mcs = [Chem.MolFromSmarts(x.smartsString) for x in (f1,f2,f3)]

終わりに
分子が類似しているということは,構造に共通点があるということを示唆します.今回は入力構造の部分構造検索から始まり,一致した構造の削除・置換や,分子の主鎖・側鎖への分離などを扱いました.
さらに指定する入力構造なしに分子の骨格を取得したり,複数分子から共通構造を取り出すことが可能であることを見てきました.今回の内容は今後取り扱う化合物のクラスタリングなどの分析に重要になってきますので,しっかりと理解を深めておきましょう.
次回はこれまでとは逆に「分子の類似度が低い」ということの意味からはじめて,ケミカルスペース中での分子間の距離や,どのように多様性の高いライブラリを構築していくかという内容を扱っていこうと思います.
>>次の記事:「ケモインフォマティクスで多様なライブラリーを構築する」
コメント