創薬化学研究では合成・試験される化合物のほとんどは医薬品にはなりません.そのため研究開発の成功率をあげるために,実際に医薬品になる化合物がどのような特徴を有するかを明らかとすべく多くの試みがなされてきました.
このような「薬らしさ(ドラッグライクスネス)」について,これまで本ブログでも何度か取り上げてきました.例えば,「色々な薬らしさの指標」という記事ではドラッグライクスネスを評価する指標として様々な基準を紹介しています.
それではこれまで承認されて実際に医薬品になった化合物はどのように調べることができるでしょうか?今回はそのような用途で,無料で利用可能な「DrugBank」とうデータベースを紹介します.
そのうえで,代表的な記述子を用いて「医薬品の占めるケミカルスペース」を可視化し,それがよくある有機化学反応の生成物とは異なる位置を占めることを確認します.
またより医薬品らしい化合物を得るためにはどのような基質(ビルディングブロック)を用いるべきかという問いに対して,Merck社の化学者によって考案された「Chemistry Informer Library」について説明します.
“Chemistry informer libraries: a chemoinformatics enabled approach to evaluate and advance synthetic methods” Chem. Sci. 2016, 7, 2604.
DrugBankとは
DrugBankはカナダのアルバータ大学で管理・運営されています.(David Wishartの研究グループ).
DrugBankデータベースは医薬品及び医薬候補化合物とそれらターゲットに関するオンラインデータベースで,2020年2月現在の最新版は2020年1月3日にリリースされたversion 5.1.5になります.全13490エントリーの内訳は以下の表になります.
| クラス | エントリー数 |
|---|---|
| 全エントリー | 13490 |
| 低分子医薬品 | 2636 |
| バイオ医薬品 | 1365 |
| 機能性食品 | 131 |
| 探索段階化合物 | >6350 |
例えばファビピラビル(アビガン)を「Avigan」で検索してみましょう.

今回は上の方に注が出ており,情報の更新が終わっていない旨の説明がでています.情報としては名前・別名,化合物の構造タイプ,構造式などの他,様々な記述子の計算値も掲載されています.

このようにオンラインでの利用は自由に利用できますが,データのダウンロードには無料の会員登録が必要になっています.
今回の記事では「STRUCTURES」から「All (3D)」というデータをダウンロードして使ってみます.

主成分分析を用いてドラッグライクケミカルスペースを可視化
Chemistry Informer Libraryとは
Merck社の化学者たちは,
- 通常の有機化学反応のスコープで出てくる化合物は脂溶性が高く,ドラッグライクネスではない
- 創薬化学者が興味のあるような基質に適用すると反応が進行しないことが多い
と述べ,彼らが論文のスコープに含まれていたら望ましいと考える基質として
- 市販のピナコールボロン酸エステルから24個
- 社内ライブラリーから芳香族ハロゲン化物を18個
を示しています.その反応が創薬化学にどの程度有用かを評価するための基質という位置づけで.「Chemistry Informer Library」と呼んでいます.
構造としては以下のようにへテロ環を中心とした化合物になります.


なおSigma-Aldrichで売られているものについては以下のように特集ページから辿ることができます.

主成分分析に用いる分子記述子
上記のChemical Scienceの論文のSupporting Informationによると以下の14個の記述子を用いたようです.多くの記述子はRDKitによってすぐに計算可能です.
| 記述子 | RDKitのメソッド |
|---|---|
| MW | Descriptors.MolWt |
| PSA | AllChem.CalcTPSA Descriptors.TPSA |
| number of atoms | Mol.GetNumAtoms |
| number of positive atoms | なし |
| number of negative atoms | なし |
| number of rotatable bonds | AllChem.CalcNumRotatableBonds Descriptors.NumRotatableBonds |
| number of rings | AllChem.CalcNumRings |
| number of aromatic rings | AllChem.CalcNumAromaticRings Descriptors.NumAromaticRings |
| number of ring assemblies | なし |
| number of stereo atoms | Chem.FindMolChiralCenters AllChem.CalcNumAtomStereoCenters AllChem.CalcNumUnspecifiedAtomStereoCenters |
| number of hydrogen bond acceptors | AllChem.CalcNumHBA Descriptors.NumHAcceptors |
| number of hydrogen bond donors | AllChem.CalcNumHBD Descriptors.NumHDonors |
| Fsp3 | AllChem.CalcFractionCSP3 Descriptors.FractionCSP3 |
| AlogP | Descriptors.MolLogP |
以下では,すぐに計算するメソッドが提供されていない3つの記述子を求める関数の実装と,これまで取り扱ったことのない「立体中心」に関するメソッドの解説をしていきます.
必要なライブラリのインポート
まずは必要なライブラリのインポートをしておきましょう.
from rdkit import rdBase, Chem from rdkit.Chem import AllChem, Draw, Descriptors, PandasTools from rdkit.Chem.Draw import IPythonConsole import numpy as np import pandas as pd import matplotlib as mpl import matplotlib.pyplot as plt import seaborn as sns sns.set() %matplotlib inline print(rdBase.rdkitVersion) # 2019.09.3 print(np.__version__) # 1.17.4 print(pd.__version__) # 0.25.3
RDKitを用いて各原子のGasteiger電荷を計算する
「RDKitの類似度マップを用いて原子ごとの寄与を可視化する」という記事でも取り上げたように,RDKitにはGasteiger電荷を計算するメソッドが実装されています.
計算された値は各Atomオブジェクトごとに「_GasteigerCharge」というプロパティで格納されます.この値の正負を判断することで,ポジティブ・ネガティブ原子の数を数え上げることとします.なおプロパティは文字列として保存されていますので,取り出す際にfloat型へと変換してあげる必要があります.
def get_num_charged_atoms(mol):
mol_h = Chem.AddHs(mol)
Chem.rdPartialCharges.ComputeGasteigerCharges(mol_h)
positive = 0
negative = 0
for atom in mol_h.GetAtoms():
if float(atom.GetProp('_GasteigerCharge')) >= 0:
positive += 1
else:
negative += 1
return positive, negative
python関数のベクトル化
上で実装した関数ではRDKitのMolオブジェクト1つに対してポジティブ・ネガティブ原子の数を返すだけで,Molオブジェクトのリストを引数に取ることはできません.この関数をベクトル化できれば,Molオブジェクトを含んだpandasのSeriesに対して一気に扱えるようになり便利です.こういった関数の変換を行うのがnumpyのfrompyfuncです.
np_get_num_charged_atoms = np.frompyfunc(get_num_charged_atoms, 1, 2)
これでMolのリストを引数に取り,ポジティブとネガティブの原子数を格納したnp.arrayを返す関数が作成できました.
リングアセンブリーの数を計算する
そもそも「ring assembly」という単語をほとんど見たことがなかったため,どのような部分構造を意味するのか最初はわかりませんでした.
調べてみたところACSの「Nomenclature of Organic Compounds」という書籍によると,「ring assembly」とは以下のように定義されるようです(強調・訳は引用者による).
意訳:「リングアセンブリー」という用語は,2またはそれ以上(縮環)の環が直接単結合または二重結合で連結された環状部分構造をいう.例えばベンゼン環が単結合で連結したビフェニルやシクロヘキサン2つが二重結合で連結した構造は「リングアセンブリー」である.一方でベンゼン環が2つの単結合で連結した構造は(3環性の)「縮環」化合物となる
<出典:Nomenclature of Organic Compounds>
以上の定義を踏まえて分子内の「ring assembly」の数を数える関数を実装してみます.いくつか方法があると思いますが,今回は
- 環を形成する原子に重複がない環のペア(縮環構造ではない)に対して
- 一方の環を構成する原子の隣接原子が,他方の環に含まれるか(環同士を繋ぐ結合があるか)
を調べることとします.
環構造に関する情報はMol.RingInfoで容易に得られます.また隣接原子についてもAtom.GetNeighborsで参照可能です.
def get_assembled_ring(mol):
ring_info = mol.GetRingInfo()
num_ring = ring_info.NumRings()
ring_atoms = ring_info.AtomRings()
num_assembled = 0
for i in range(num_ring):
for j in range(i+1, num_ring):
x = set(ring_atoms[i])
y = set(ring_atoms[j])
if not x.intersection(y): # 2つの環が縮環でない場合に
for x_id in x:
x_atom = mol.GetAtomWithIdx(x_id)
neighbors = [k.GetIdx() for k in x_atom.GetNeighbors()]
for x_n in neighbors:
if x_n in y: # 環同士を繋ぐ結合があるか否か
num_assembled += 1
return num_assembled
分子中の不斉炭素の数
AllChem.CalcNumAtomStereoCenters(mol)
AllChem.CalcNumUnspecifiedAtomStereoCenters(mol)
FindMolChiralCentersは分子中の立体中心の数を返します.DrugBankのデータは立体構造が含まれていますが,念のためincludeUnassigned=Trueにしておくと立体が明示的になっていないものも含めて数えることが可能です.
別々に計算したい場合に利用可能なメソッドもAllChemに用意されています.今回はこちらを使うことにします.
def get_num_stereocenters(mol):
return AllChem.CalcNumAtomStereoCenters(mol) + AllChem.CalcNumUnspecifiedAtomStereoCenters(mol)
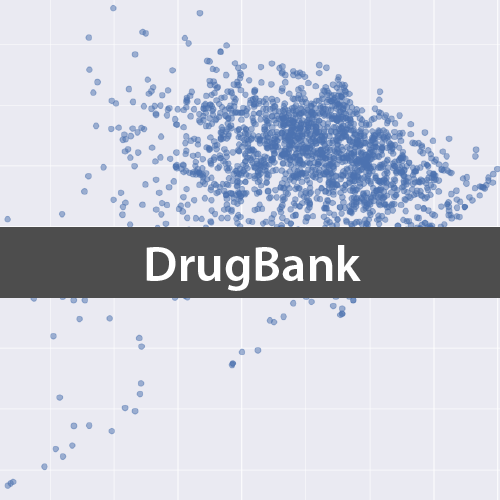
DrugBank化合物を用いて主成分分析
それではDrugBankからダウンロードしたSDFを用いて,承認済み医薬品の記述子を計算し,主成分分析を行うことで2次元に化合物を可視化してみます.
DrugBankではSDFの「DRUG_GROUPS」という項に「approved」が含まれていれば承認済み化合物ですので,pandas.queryを用いてスクリーニングしていきます.
以下のコードでは
- 記述子を計算する関数を定義
- SDFを読み込み,承認済み医薬品のみに絞り込み,記述子計算
- 記述子の標準化と主成分分析
- 散布図を用いて可視化
- 主成分の累積寄与率を表示
# 1. Molオブジェクトを含むデータフレームから記述子を計算する関数
def calc_descriptors(dataframe):
dataframe['MW'] = dataframe.ROMol.map(Descriptors.MolWt)
dataframe['PSA'] = dataframe.ROMol.map(Descriptors.TPSA)
dataframe['n_atoms'] = dataframe.ROMol.map(lambda x: x.GetNumAtoms())
dataframe['n_positive'], dataframe['_n_negative'] = np_get_num_charged_atoms(dataframe.ROMol)
dataframe['n_rot_bonds'] = dataframe.ROMol.map(Descriptors.NumRotatableBonds)
dataframe['n_rings'] = dataframe.ROMol.map(AllChem.CalcNumRings)
dataframe['n_ar_rings'] = dataframe.ROMol.map(Descriptors.NumAromaticRings)
dataframe['n_ring_asmbl'] = dataframe.ROMol.map(get_assembled_ring)
dataframe['n_stereo'] = dataframe.ROMol.map(get_num_stereocenters)
dataframe['n_HBA'] = dataframe.ROMol.map(Descriptors.NumHAcceptors)
dataframe['n_HBD'] = dataframe.ROMol.map(Descriptors.NumHDonors)
dataframe['Fsp3'] = dataframe.ROMol.map(Descriptors.FractionCSP3)
dataframe['logP'] = dataframe.ROMol.map(Descriptors.MolLogP)
# 2. SDFの読み込みと化合物のスクリーニング
df = PandasTools.LoadSDF('./3D structures.sdf', removeHs=False)
df_approved = df.query('DRUG_GROUPS.str.contains("approved")', engine='python')
calc_descriptors(df_approved)
print(len(df_approved)) # 2017
# 3. 記述子の標準化と主成分分析
from sklearn.decomposition import PCA
from sklearn.preprocessing import StandardScaler
descs = ['MW', 'PSA', 'n_atoms', 'n_positive', '_n_negative', 'n_rot_bonds',
'n_rings', 'n_ar_rings', 'n_ring_asmbl', 'n_stereo', 'n_HBA', 'n_HBD',
'Fsp3', 'logP']
scalar = StandardScaler()
scalar.fit(df_approved[descs])
df_std = scalar.transform(df_approved[descs])
pca = PCA(n_components=4)
pca.fit(df_std)
df_std_trans = pca.transform(df_std)
# 4. 可視化
fig = plt.figure(figsize=(12,12))
ax = fig.add_subplot(111)
ax.scatter(-df_std_trans[:,0], -df_std_trans[:,1], s=50, alpha=0.5)
ax.set_xlabel('PCA 1')
ax.set_ylabel('PCA 2')
# 5. 主成分の累積寄与率
np.cumsum(pca.explained_variance_ratio_)

array([0.42350166, 0.64951455, 0.78629754, 0.88027908])
計算した主成分の累積寄与率がやや異なりますが,計算しているソフトの違いと考えられます.また本質的には何も変わりませんが論文中のグラフと似せるために,両主成分の値を反転(-1を掛ける)させています.
鈴木カップリング生成物のドラッグライクスネスケミカルスペースへの可視化
論文では鈴木らによるカップリング反応の最初の報告(Syn. Commun. 1981, 11, 513.)の生成物を用いて,同じように記述子の計算,主成分分解,マッピングを行っています.生成物の構造は以下の通り,シンプルなビアリール構造です.

suzuki = PandasTools.LoadSDF('./Suzuki.sdf')
suzuki['ROMol'] = suzuki.ROMol.map(Chem.AddHs)
calc_descriptors(suzuki)
suzuki_std = scalar.transform(suzuki[descs])
suzuki_std_trans = pca.transform(suzuki_std)
fig = plt.figure(figsize=(12,12))
ax = fig.add_subplot(111)
ax.scatter(-df_std_trans[:,0], -df_std_trans[:,1], s=50, alpha=0.5, label='DrugBank')
ax.scatter(-suzuki_std_trans[:,0], -suzuki_std_trans[:,1], s=100, alpha=0.5, label='Seminal Suzuki')
ax.set_xlabel('PCA 1')
ax.set_ylabel('PCA 2')
plt.legend()

論文通り,随分と医薬品とは異なる部位を占めることがわかります.
Informer Libraryを用いた主成分分析
最後にInformer Libraryを用いて発生させた化合物に対して,同じように主成分分析を行い図示してみましょう.その際,論文とは異なりピナコールボロン酸エステルとハロゲン化物を直接カップリングさせた化合物を用いてみます.
以下のコードでは,
- SDFの読み込み
- クロスカップリング反応の定義と実行
- 記述子の計算,標準化,主成分分析
- 可視化
と行っています.
なおカップリング反応の定義ではハロゲン化物が
- ヨウ素
- 臭素
- 塩素
の場合があるため,反応性の高い順番に反応式を定義しています.
また反応実行にあたって1つの臭化物(ArBr[12])を用いた際に,その後の記述子計算でエラーが頻発したことから除去しています.
# 1. SDF読み込み
suppl1 = Chem.SDMolSupplier('./ArBr.sdf')
ArBr = [x for x in suppl1 if x is not None]
suppl2 = Chem.SDMolSupplier('./Bpin.sdf')
Bpin = [x for x in suppl2 if x is not None]
# 2. 反応の定義
def gen_coupling_pattern(mol):
formula = AllChem.CalcMolFormula(mol)
if 'I' in formula:
halogen = 'I'
elif 'Br' in formula:
halogen = 'Br'
else:
halogen = 'Cl'
return '[c:1]{}.[c:2](B(O)O)>>[c:1][c:2]'.format(halogen)
def Suzuki_Rxn(halide, boronate):
rxn = AllChem.ReactionFromSmarts(gen_coupling_pattern(halide))
product = rxn.RunReactants([halide, boronate])
if len(product):
p = product[0][0]
return p
else:
return None
# 反応の実行
products = []
for boronate in Bpin:
for halide in ArBr[:12]+ArBr[13:]:
products.append(Suzuki_Rxn(halide, boronate))
print(len(products)) # 408 = 24*17
for m in products:
AllChem.Compute2DCoords(m)
# 3. 記述子計算,標準化,主成分分析
df_library = pd.DataFrame(products, columns=['ROMol'])
df_library.ROMol = df.ROMol.map(Chem.AddHs)
calc_descriptors(df_library)
df_library_std = scalar.transform(df_library[descs])
df_library_std_trans = pca.transform(df_library_std)
# 4. 可視化
fig = plt.figure(figsize=(12,12))
ax = fig.add_subplot(111)
ax.scatter(-df_std_trans[:,0], -df_std_trans[:,1], s=50, alpha=0.5, label='DrugBank')
ax.scatter(-suzuki_std_trans[:,0], -suzuki_std_trans[:,1], s=100, alpha=0.5, label='Seminal Suzuki')
ax.scatter(-df_library_std_trans[:,0], -df_library_std_trans[:,1], s=100, alpha=0.5, label='Informer Library')
ax.set_xlabel('PCA 1')
ax.set_ylabel('PCA 2')
plt.legend()

少なくとも今回用いた記述しに関しては,ほとんどDrugBankと重なっています.
終わりに
今回は「DrugBankは承認済み医薬品のデータベース:主成分分析によるドラッグライクケミカルスペースの可視化」という話題について,
- DrugBankデータベースとは何か
- RDKitを用いた記述子計算法の復習
- 主成分分析を用いたドラッグライクケミカルスペースの可視化
- 薬らしい化合物群にアクセスするための「Informer Library」
などについて触れました.
創薬化学研究の目的は「医薬品」を世に送り出すことですので,承認済み医薬品が集められたDrugBankは有用なデータベースと言えます.例えば,既知の生理活性物質を集めたChemblからランダムに抽出した化合物群とDrugBankと一緒に可視化した図などが論文でよく見られます.
コメント