「RDKitでRECAPを用いた分子のフラグメント化」という記事では,医薬品様化合物のフラグメント化方法としてRECAPと呼ばれる方法を扱いました.今回は同様に分子のフラグメント化アルゴリズムであるBRICS(Breaking of Retrosynthetically Interesting Chemical Substructures)について扱います.
いずれの方法も
- (生理活性を有する)化合物を
- 逆合成の観点も含めて
フラグメント化するという点ではRECAPとBRICSは類似していますが,RDKitにおける2つの実装はかなり異なっています.今回はその違いに焦点を当てながら見ていきたいと思います.
BRICS
RECAPでは11種類のフラグメントを考えて切断しましたが,BRICSではさらに切断箇所を増やして16種類のフラグメントを考えます.そのため,同じ化合物ライブラリーに対して2つの手法を別個に施した際には,必然的にRECAPと比べると
- 切断されない分子数の減少
- フラグメント総数の増加
が,BRICS適用群では見られます.
RDKitにおけるBRICSの実装
「RDKitでRECAPを用いた分子のフラグメント化」という記事で解説したように,RDKitでRECAPを分子に適用すると,フラグメント化の工程がツリー構造になって得られます.
BRICSも分子を一定のルールに則ってフラグメント化するアルゴリズムですが,RDKitではツリー構造ではなく,RECAPのLEAFに相当するフラグメントの集合が得られます.以下で,具体的に見ていくことにしましょう.
分子の読み込み
公式ドキュメント「Getting Started with the RDKit in Python: BRICS Implementation」の例がCDK2になっていますので,ここでは同様にキナーゼ阻害剤としてZINC15データベースより取得したJAK3阻害剤を使ってみます.なんとなく似たような平面構造が多くなりそうな気がすると思います.
まずは必要なライブラリのインポートと分子の読み込みをします.
from rdkit import rdBase, Chem
from rdkit.Chem import AllChem, Draw
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import BRICS
print(rdBase.rdkitVersion) ### 2018.09.1
### 分子の準備
suppl = Chem.SDMolSupplier('JAK3-usual.sdf')
mols = [x for x in suppl if x is not None]
len(mols) ### 100
全部で100分子のうち,最初の分子は以下のような構造でした(3次元構造のままだと見にくいので2次元に書き直してあります).

BRICSによるフラグメント化
Chem.BRICS.BreakBRICSBonds(mol)
RDKitでBRICSによるフラグメント化を実施するにはBRICSDecomposeを用います.デフォルト設定では切断位置にダミーアトムを挿入したSMILESのsetが得られます.
frag = BRICS.BRICSDecompose(mols[0]) len(frag) ### 6
{'[1*]C(=O)[C@H]([4*])C(C)(C)C',
'[1*]C([6*])=O',
'[14*]c1cnc2[nH]cc([16*])c2n1',
'[15*]C1CC1',
'[5*]N1CC(C#N)C1',
'[5*]N[5*]'}
一方でreturnMolsオプションをTrueにすることで,MOLオブジェクトが直接得られます.ただし分子内の座標を保持したままですので,下の例のようにそのままの描画には向きません.
f_mols = BRICS.BRICSDecompose(mols[0], returnMols=True) Draw.MolsToGridImage(f_mols, molsPerRow=3)

BreakBRICSBondsメソッドでは切断部分をマークします.切断箇所を示すフラグからMOLオブジェクトを構築したい場合には,「RDKitを用いた部分構造検索とMCSアルゴリズム」という記事でもみたようにGetMolFragsメソッドを用います.
m = BRICS.BreakBRICSBonds(mols[0]) print(Chem.MolToSmiles(m)) fm = Chem.GetMolFrags(m, asMols=True)
[1*]C(=O)[C@H]([4*])C(C)(C)C.[1*]C([6*])=O.[14*]c1cnc2[nH]cc([16*])c2n1.[15*]C1CC1.[5*]N1CC(C#N)C1.[5*]N[5*]
得られたフラグメントは今回も3次元座標を維持したままですので,直接の描画には向きません.また得られるフラグメントの順番が異なっていることがわかると思います.

フラグメント頻度のカウントと全フラグメント数
続いて100分子全てに対してBRICSを適用していき,標準ライブラリーcollectionsにあるCounterを用いて,各フラグメントの回数を調べてみることにしましょう.
from collections import Counter
cnt = Counter()
for mol in mols:
pieces = BRICS.BRICSDecompose(mol)
for p in pieces:
cnt[p] += 1
len(cnt) ### 158
100分子から,158個のフラグメントが得られました.出現頻度別にヒストグラムを描いてみましょう.
import matplotlib as mpl
import matplotlib.pyplot as plt
with plt.style.context('seaborn'):
plt.hist(cnt.values(), bins=range(max(cnt.values())+1))
plt.xlabel('freq of fragment')

ほとんどのフラグメントは1,2回しか登場しませんが,いくつかのフラグメントが優先して出てきているようです.ここでは上位9個程度の構造を見てみましょう.

2級・3級アミンやカルボニルが圧倒的に上位ですが,シクロプロピルやへテロ芳香族などの特徴的な部分構造が上位に入ってきているのがわかります.
このようにあるライブラリ中でよく用いられる部分構造を眺めたりすることが可能です.
BRICSを用いたバーチャルライブラリーの構築
BRICSDecomposeで得られたフラグメントのMOLオブジェクトを部分構造として利用することで仮想ライブラリーの構築が簡単に行えます.BRICSBuildメソッドは,作成したバーチャルライブラリー中の分子を生成するpythonのジェネレーターを返します.
ジェネレーターからは組み込み関数nextを使うことで分子を取り出すことができます.以下のコードでは,
- 全てのフラグメントを集める
- SMILESからMOLオブジェクトの作成
- BRICSBuildを用いてジェネレーターの生成
- 分子を1000個作成し,ランダムにシャッフル
という作業を行っています.作成された分子はUpdatePropertyCacheを用いて価数などの情報を更新しておいた方がいいようです.
### フラグメント集合を作成
allfrags = set()
for mol in mols:
frag = BRICS.BRICSDecompose(mol)
allfrags.update(frag)
len(allfrags) ### 158
### smilesをMOLオブジェクトに変換し,ジェネレーターを作成
allcomponents = [Chem.MolFromSmiles(f) for f in allfrags]
builder = BRICS.BRICSBuild(allcomponents)
type(builder) ### generator
### 分子1000個を作成し,シャッフル
generated_mols = []
for i in range(1000):
m = next(builder)
m.UpdatePropertyCache(strict=True)
generated_mols.append(m)
import numpy as np
np.random.seed(1234)
np.random.shuffle(generated_mols)
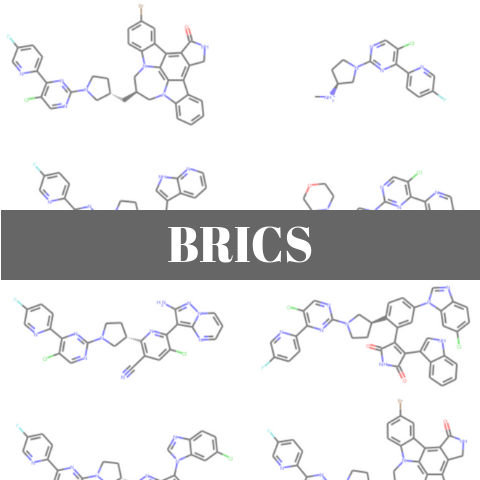
Draw.MolsToGridImage(generated_mols[:9], molsPerRow=3, subImgSize=(300,150))
シャッフル後の最初の9個の分子の構造は以下のようでした.アミンがカチオンになっていたりする点が気になりますが,先ほど見たような部分構造の組み合わせによって仮想ライブラリーを構築することができました.

RECAPとBRICSの比較
最後にRECAPとBRICSの比較を簡単に行ってみます.
まずは得られるフラグメント数です.RECAPでは以下のコードのように140個のフラグメントが得られました.BRICSでは158でしたから,やや少なくなっています.
all_leaves = set()
for mol in mols:
tree = Recap.RecapDecompose(mol)
leaves = tree.GetLeaves().keys()
all_leaves.update(leaves)
len(all_leaves) ### 140
続いて実行速度を見てみます.手元のMacBookProでは10%ほどRecapが速かったです.
def recap_frag_smiles(mols):
all_leaves=set()
for mol in mols:
tree = Recap.RecapDecompose(mol)
leaves = tree.GetLeaves().keys()
all_leaves.update(leaves)
return all_leaves
def brics_frag_smiles(mols):
all_frags = set()
for mol in mols:
frags = BRICS.BRICSDecompose(mol)
all_frags.update(frags)
return all_frags
%timeit recap_frag_smiles(mols)
%timeit brics_frag_smiles(mols)
588 ms ± 3.77 ms per loop (mean ± std. dev. of 7 runs, 1 loop each) 652 ms ± 5.89 ms per loop (mean ± std. dev. of 7 runs, 1 loop each)
なおBRICSDecomposeとBreakBRICSBondsでは後者の方が圧倒的に速いです.
%timeit [BRICS.BRICSDecompose(mol, returnMols=True) for mol in mols]
def brics_bond_mol(mol):
fragments = BRICS.BreakBRICSBonds(mol)
f_mols = Chem.GetMolFrags(fragments, asMols=True)
return f_mols
%timeit [brics_bond_mol(mol) for mol in mols]
647 ms ± 2.77 ms per loop (mean ± std. dev. of 7 runs, 1 loop each) 163 ms ± 1.17 ms per loop (mean ± std. dev. of 7 runs, 10 loops each)
終わりに
今回は分子のフラグメント化アルゴリズムの1つであるBRICSについて学んできました.BRICSはRECAPと同様に,
- 化合物のフラグメント化によって多数の部分構造を与えるアルゴリズムであること
- RDKitを用いればBRICSの実行が容易であること
- バーチャルライブラリーの構築がコマンド1つで実行可能であること
などが理解して頂けたと思います.
一連の化合物ライブラリーをもとにして,どのようにして類似骨格を有する化合物を取得するかを扱ってきました.RECAPもBRICKも逆合成的な観点を含んだフラグメント化ではありますが,どれだけ有望そうな化合物がin silicoでデザインできたとしても,実際に合成できなければ意味がありません.
次回は,化合物の「合成容易さ」という指標をどのように定量化するかについて説明していきたいと思います.
>>次の記事:「RDKitで合成難易度を評価して化合物をスクリーニング」
コメント