「RDKitとscikit-learnで機械学習:変異原性をk-最近傍法で予測」という記事では,まず「教師あり学習」と「教師なし学習」,「回帰」と「分類」といった基本的な機械学習の用語を学びました.
その後,最も単純な機械学習手法とも言われるk-最近傍法を用いて,RDKitとscikit-learnを使ったモデル構築を行いました.
モデル構築にあたっては,
- 分子の情報をどのように入力ベクトルとしてエンコードするかによって同じ機械学習手法(k-最近傍法)を用いても精度が変わること
- 訓練データについて精度が高いモデルが必ずしもテストデータでよい精度を上げるわけではないこと(過剰適合)
を確認しました.これらの項目はこれから機械学習の理解を深めていく上で大切になります.
今回は前回と同じAmes試験の結果を予測するモデルを「決定木」と呼ばれる手法によって行っていきます.機械学習モデルはよく中身のわからない「ブラックボックス」と言われますが,決定木で作成したモデルは人間が理解しやすいものになります.
「Benchmark Data Set for in Silico Prediction of Ames Mutagenicity」J. Chem. Inf. Model. 2009, 49, 2077. (DOI: 10.1021/ci900161g)
決定木とは
決定木とは「yes」「no」で答えられる質問を繰り返しながら,階層構造を作成していく方法になります.トップダウンとボトムアップのいずれの方法でも階層構造が作れますが,現在はトップダウン的に決定木を作成するのが主流です.
「質問」を作成するにはある特徴量に着目して,2つに分割するルールを作成します.具体例として以下に示すような二次元に分布する赤と青の5つの点を考えてみます.

最初の質問(a)では
- x座標が4より大きいか?
について考えます.すると2つの赤い点とその他の3点に分類されます.後者に対して次の質問(b)
- y座標が2より大きいか?
を考えます.すると2つの青い点と1つの赤い点に分類され,全ての点を分類できました.一連の流れを図にすると下に示すような階層構造ができあがります.

先にも述べたようにこのような階層構造を眺めることで,モデルがどのようなルールに基づいてデータセットを分類していったかが理解できます.
決定木におけるノード分割規則
決定木において,質問の取り方はx座標,y座標に対してそれぞれ4(5-1)通り存在します.階層構造を組み立てるにあたっては全ての分割方法を考慮し,「不純度」とよばれる評価関数を用いて選択します.
評価関数としては
- ノードにおける誤り率
- 交差エントロピー
- ジニ係数(Gini index)
などが用いられます.
scikit-learnにおける決定木の実装
決定木はscikit-learnではsklearn.treeに実装されています.なお評価関数(criterion)としては交差エントロピー(”entropy”)とジニ係数(”gini”; 標準設定)が実装されています.
分子の読み込み
まずはデータセットから分子を読み込み,分子の情報を入力ベクトルとして変換しましょう.
論文のデータはタブ区切りのテキストファイルで,「SMILES」「CAS NO」「変異原性」の3つのデータが6512化合物について格納されています.
以下のコードでは
- 必要なライブラリのインポート
- pandasのデータフレームとして情報を読み込む
- SMILESからRDKitのMOLオブジェクトを構築
- 読み込めないSMILESを含むデータを除去
- MACCSKeyフィンガープリントを入力ベクトルとして準備
という順番で処理を行っています.
## 1. 必要なライブラリのインポート
from rdkit import rdBase, Chem
from rdkit.Chem import AllChem, Draw, PandasTools, Descriptors
from rdkit.Chem.Draw import IPythonConsole
import matplotlib as mpl
import matplotlib.pyplot as plt
import seaborn as sns
import pandas as pd
import numpy as np
mpl.style.use('seaborn')
print(rdBase.rdkitVersion) # 2018.09.1
## 2. pandasデータフレームに分子を読み込み
df = pd.read_csv('./ci900161g_si_001/smiles_cas_N6512.smi', header=None, sep='\t')
df.columns = ['smiles', 'CAS_NO', 'activity']
## 3. SMILESからRDKitのMOLオブジェクトを構築
PandasTools.AddMoleculeColumnToFrame(frame=df, smilesCol='smiles')
## 4. 読み込めない分子を削除
df['MOL'] = df.ROMol.map(lambda x: False if x == None else True)
del_index = df[ df.MOL == False].index
df2 = df.drop(del_index)
## 5. フィンガープリントを準備
maccskeys = []
for m in df2.ROMol:
maccskey = [x for x in AllChem.GetMACCSKeysFingerprint(m)]
maccskeys.append(maccskey)
maccskeys = np.array(maccskeys)
maccskeys.shape # (6506, 167)
決定木を利用したモデル構築
model_selection.train_test_split(X, y)
scikit-learnの決定木は何もパラメーターを指定しないと,与えられた訓練データを用いてどんどんと木を伸長させていきます(枝を増やします).まずは何も制限を設けずにモデルを作成してみましょう.
from sklearn.tree import DecisionTreeClassifier
from sklearn.model_selection import train_test_split
X_train, X_test, y_train, y_test = train_test_split(maccskeys, df2.activity, random_state=0)
tree = DecisionTreeClassifier(random_state=0)
tree.fit(X_train, y_train)
print('accuracy on train set: {:.3f}'.format(tree.score(X_train, y_train)))
print('accuracy on test set: {:.3f}'.format(tree.score(X_test, y_test)))
訓練データに対してテスト用データの精度が随分と劣ることが見てとれます.これは過剰適合を示唆していますので,続いてモデルの複雑度を制御することで過剰適合を抑えてみましょう.
accuracy on train set: 0.976 accuracy on test set: 0.750
決定木の調整法
決定木においてモデルの複雑さを制御する方法には,
- 木を成長させた後で情報の少ない枝を除く(事後枝刈り;post-pruning)
- 木の成長を早い段階で止める(事前枝刈り;pre-pruning)
という2つの方法があります.scikit-learnには後者の事前枝刈りが実装されています.
事前枝刈りとしては具体的には
- 木の深さを制限する:max_depth
- 葉の最大値を制限する:max_leaf_nodes
- 新しくできる葉で除かれれる不純物の割合:min_impurity_decrease
という3種類が実装されています.いずれか1つを用いるのみで十分です.
以下のコードではmax_depthを制御することで,どのようにテストセットの精度が変化するかを見ていきます.
accs_train = []
accs_test = []
for i in range(1,21):
tree_i = DecisionTreeClassifier(max_depth=i, random_state=0)
tree_i.fit(X_train, y_train)
acc_train = tree_i.score(X_train, y_train)
acc_test = tree_i.score(X_test, y_test)
accs_train.append(acc_train)
accs_test.append(acc_test)
今回の場合はmax_depth=9が最も精度がよさそうです.

決定木の可視化方法
決定木は中身が人間にも理解しやすい機械学習モデルであると述べました.ここでは決定木を可視化する方法について説明します.
大きな深いモデルでは細かすぎますので,簡単のためにmax_depthを2に設定した小さめのモデルを用いていきます.
tree2 = DecisionTreeClassifier(max_depth=2) tree2.fit(X_train, y_train)
graphvizを用いた可視化
決定木の可視化方法として最も広く使われている方法がgraphvizを用いたものです.graphvizとはその名前が示唆するようにグラフ構造を描画するためのソフトウェアになります.描画には特別な文法が必要となりますが,幸いなことにscikit-learnではgraphvizが読み込める「dot」形式で決定木をエクスポートする機能が搭載されています.
graphvizのインストール
インストールには
- graphvizのインストール
- pythonでgraphvizを利用する準備
の2段階で行います.まずはmacならhomebrew,linuxならapt-getやyumでインストールし,その後pytonで利用するためにpipでインストールします.
brew install graphviz pip install graphviz
dotファイルの作成
export_graphvizではいくつかの引数を取ります.代表的なものとしては下記のものがあります.
| 引数 | 説明 |
|---|---|
| dicision_tree | 決定木のモデル |
| out_file | 作成するdotファイル名 |
| class_names | 分類クラスの名前 |
| feature_names | 特徴量の名前 |
| filled | Trueでどちらのクラスに分類されているか色分け |
| rounded | Trueでノードの角を丸める |
以下のコードでは一旦dotファイルを作成し,それを読み込んでいます.またfilled=Trueとすることでノードに色をつけています.
from sklearn.tree import export_graphviz
export_graphviz(tree2, out_file='tree2.dot', class_names=['negative', 'positive'],
feature_names=range(1,168), filled=True)
import graphviz
with open('tree2.dot') as f:
dot_graph = f.read()
graphviz.Source(dot_graph)
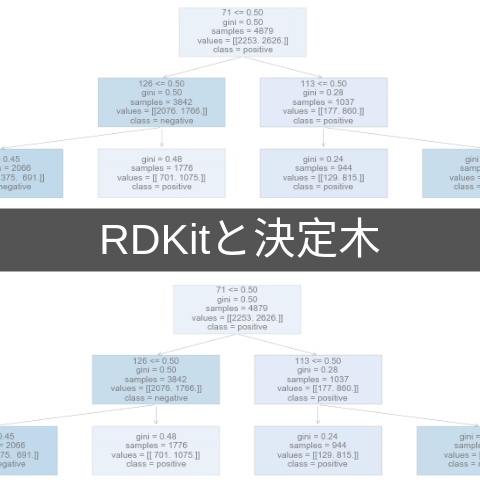
今回の場合「陽性」の多いノードが青色で,「陰性」の多いノードは橙色で表されていることがわかります.またサンプル数,判別条件,gini係数などの基本情報も記されていますので,どのようにモデルが分類を進めていったのかを読み解くことが可能です.

以下のコードではノードに色をつけずに,rounded=Trueとすることで丸みをつけています.
dt = export_graphviz(tree2, out_file=None, class_names=['negative', 'positive'],
feature_names=range(1,168), rounded=True, filled=False)
graphviz.Source(dt)
同じモデルを図にしただけですが,随分と雰囲気が違いますね.

dtreepltを用いた可視化
scikit-learnのDecisionTreeClassifierオブジェクトには決定木を可視化するための全ての情報が含まれています.そのためgraphvizのような外部のソフトウェアに頼らずにpython内で完結することを目指し,matplotlibを用いて可視化するために開発されたライブラリーがnekoumei氏によるdtreepltです.
dtreepltのインストール
pipでインストールします.
pip install dtreeplt
dtreepltを使った可視化
dt2 = dtreeplt(model=tree2, feature_names=range(1,168), target_names=['negative', 'positive'])
view = dt2.view()
view.savefig('./tree2.png', transparent=True)
同じモデルを用いて可視化していますので,当然中身はgraphvizと同じになります.今後は以下に示すdtreevizのような表現をmatplotlibを用いて実装することを目指しているようです.

dtreevizを用いた可視化
graphvizを用いて得られる図はモデルの中身を理解するのに必要な情報は集まっています.一方で文字情報が多いため,「可視化」という観点からはやや弱いです.こういった問題を解決するために作成されたのが,Terence ParrとPrince Groverの両氏によるdtreevizというライブラリーになります.サンプル画像を見てみるとわかりますが,非常にきれいに可視化されています.
日本語の記事ですと例えば「決定木の可視化ライブラリ「dtreeviz」が凄かったのでまとめる」あたりが参考になると思います.
dtreevizのインストール
インストールはgithubにある説明通りでうまくいくはずなのですが,手許のMacへのインストールでは下記のissueと同じ問題が出ていて個人的にはまだ解決できていません.
MacOS: Error: invalid option: –with-librsvg #33
Linux(CentOS)上へは問題なくインストールできました.
dtreevizの使い方
基本的にはgithubにあがっているJupyter notebookを眺めればわかります.
可視化の中心はdtreevizメソッドになります.他のライブラリーと同様に,dtreevizは決定木のモデル以外にもいくつかの引数を取りますので,下記に代表的なものをまとめておきます.
| 引数 | 説明 |
|---|---|
| target_name | ターゲットの名前 |
| feature_names | 特徴量の名前 |
| class_names | 分類クラスの名前 |
| fancy | ヒストグラムによる分岐点の表示を行うか.デフォルトはTrue |
| orientation | 決定木をどの方向に伸長するか.デフォルトは’TD'(top-down,上から下).’LR'(left-right,左から右) |
| histtype | ヒストグラムの種類.デフォルトは’barstacked’で,4-5種類までの分類におすすめらしい.それ以上なら’bar’を利用 |
| show_node_labels | 各ノードに番号を振る.デフォルトはFalse. |
| X | 与えたデータに対してモデルの判定を表示 |
それではいくつか具体例を見ていきましょう.
from dtreeviz.trees import dtreeviz
viz = dtreeviz(tree2, X_train, y_train, target_name='mutagenicity',
feature_names=range(1,168),
class_names=['negative', 'positive'])
display(viz)

続いて深さを3と大きくする代わりに,ヒストグラムによる図示を辞めてみます(fancy=False).
tree3 = DecisionTreeClassifier(max_depth=3) tree3.fit(X_train, y_train) viz3 = dtreeviz(tree3, X_train, y_train, target_name='mutagenicity', feature_names=range(1,168), class_names=['negative', 'positive'], fancy=False) display(viz3)

深さ3を保つながら,左から右へと木を成長させてみます(orientation=’LR’).
v = dtreeviz(tree3, X_train, y_train, target_name='mutagenicity', feature_names=range(1,168), class_names=['negative', 'positive'], orientation='LR') display(v)

今度は右から左へと木を成長させ(orientation=’RL’),ヒストグラムを重ねずに表示してみます(histtype=’bar’).
v = dtreeviz(tree3, X_train, y_train, target_name='mutagenicity', feature_names=range(1,168), class_names=['negative', 'positive'], orientation='RL', histtype='bar') display(v)

ベンツピレンに対する予測
最後に変異原性の知られるベンツピレンについてmax_depth=2のモデルで予測を行い,dtreevizを用いて判断基準を可視化してみましょう.なお訓練データには類似構造は多数存在するもののベンツピレンそのものは入っていませんでした.
まずは分子を読み込んで,部分構造で検索してみます.
m1 = Chem.MolFromSmiles('c1ccc2c(c1)cc3ccc4cccc5c4c3c2cc5')
df2[ df2.ROMol >= m1 ][:3]
df2[ df2.ROMol >= m1].activity.plot(kind='hist')
ベンツピレンを部分構造として含むものが多数存在し,その8割近くが陽性のようです.


続いてフィンガープリントを用意し,dtreevizメソッドに渡すことで可視化してみます(X=fp).さらに説明しやすいようにノードに番号を振ってみます(show_node_labels=True).
fp = [x for x in AllChem.GetMACCSKeysFingerprint(m1)]
fp = np.array(fp)
v = dtreeviz(tree2, X_train, y_train, target_name='mutagenicity',
feature_names=range(1,168),
class_names=['negative', 'positive'],
orientation='TD',
show_node_labels=True,
X=fp)
display(v)

決定木における各特徴量の重要度
決定木のモデルにおいて,各特徴量がどの程度識別に重要であるかは,feature_importances_に格納されています.
下のコードではmax_depth=9のモデルについてプロットしています.
tree9 = DecisionTreeClassifier(max_depth=9) tree9.fit(X_train, y_train) plt.plot(tree9.feature_importances_) plt.xlim(0,167)
先ほどの決定木の可視化においても明らかでしたが,71と126が重要そうであることが確認できました.気をつけるべき点として,重要度の低い特徴量が必ずしも情報量が少ないとは限らないという点が挙げられます.単純に他の特徴量と類似の情報を持っているために,今回のモデル化では採用されなかっただけかもしれません.

終わりに
今回は「scikit-learnの決定木でAmes試験データセットを機械学習」という内容について,
- 決定木とは何か
- scikit-learnにおける決定木の実装
- 決定木の可視化方法
について触れてきました.その中でどのようにモデルの複雑さを制御していくかについても学びました.決定木は
- モデルの可視化が容易
- その内容が理解しやすい
- 特徴量の前処理を必要としない
といった長所があります.一方で,
- 容易に過剰適合を起こす
- データの小さな変化でモデルが大きく変わることがある(剛健性が低い)
などといった欠点も知られています.このような欠点の多くは複数の決定木を組み合わせることで克服できることが知られています.
次回は「アンサンブル法」と呼ばれる複数の機械学習モデルを組み合わせる方法について触れ,特にランダムフォレストと呼ばれる手法について学習していきます.
>>次の記事:「RDKitでランダムフォレスト:機械学習でも「みんなの意見」は案外正しい」
コメント