化合物ライブラリー中の大量の化合物から,どれをスクリーニングにかけるかはケモインフォマティクスにおける重要な課題です.本サイトでもこれまで
- 「RDKitで薬らしさを定量的に評価する」という記事で,開発可能性が高まるような化合物の基準
- 「ケモインフォマティクスで多様な化合物ライブラリーを構築する」という記事で,ライブラリー中から多様な分子を選択するアルゴリズム
- 「RDKitで合成難易度を評価して化合物をスクリーニング」という記事で,特にin silicoで設計した化合物の合成難易度を評価する方法
といった話題について扱ってきました.通常,ハイスループットスクリーニング(HTS)では決まった濃度で次々とアッセイをしていき,その後ヒット化合物について活性の濃度依存性などを見ていくことになります.
スクリーニングにあたっては,酸ハライドのような明らかに反応性の高い官能基を有する化合物は事前に除いておきます.それでも色々な標的に対するスクリーニングにおいて一貫して「ヒット」に含まれている構造が存在することが知られてきました.そのような化合物は,我々が期待するような作用機序とは異なる機構で「活性」を示すことが多く,事前に除いておくことが偽陽性を減らすことに繋がります.
今回取り上げるPAINSフィルターはそのような部分構造をリスト化したもので,賛否両論ありながら忌避構造リストとしては有名なものになります.
PAINS化合物とは
2010年のJMCでは,PerkinElmer社のAlphaScreenを用いた6つのHTSにおけるヒット化合物の数を調べています.
| ヒットカウント | 6 | 5 | 4 | 3 | 2 | 1 | 0 | 合計 |
|---|---|---|---|---|---|---|---|---|
| 化合物数 | 362 | 785 | 915 | 1220 | 4689 | 12077 | 73164 | 93212 |
<出典:JMC 2010, 53, 2719>
上の表のように,例えば全てのアッセイで「ヒット」と判定された化合物が362ありました.著者らはこういった化合物に存在する問題となりそうな「部分構造」を,ある基準でリスト化することとしました.このような化合物(Pan-Assay INterference compoundS)から得られた480個のリストがPAINSフィルターになります.
偽陽性化合物であることを見抜けずに,その構造を最適化してしまった場合に費やされるお金と時間は文字通り「無駄」になりますので,このようなリストの登場は歓迎されました.
またこういった偽陽性化合物が活性化合物として論文に報告され,コントロール試薬として市販されてしまうことは,今後の科学の発展にも大きな障害となります.アメリカ化学会では「The Ecstasy and Agony of Assay Interference Compounds」と題した2017年の論説で,論文投稿にあたっては
- PAINSフィルタリングを行うこと
- PAINS化合物については,追加実験によって化合物の真の活性を担保すること
を強く推奨しています.以前からPAINSフィルターに関しては議論がありましたが,この論説以降さらに活発になりました.
PAINSフィルターに関する議論
PAINSフィルターに関する問題点としては,
- AlphaScreenアッセイのみを用いていること
- たった6回のタンパク質間相互作用に対するアッセイから得られていること
- 商用化合物データを使っていること
- 7割近い構造はライブラリー中でごく少数回登場しただけであること
などが挙げられています.
より大規模でオープンなデータベースであるPubChemを用いてPAINSフィルターを検証したところ,フィルタリングされた化合物の多くはAlphaScreenを用いたPPIアッセイでは頻繁にヒットとはなっていないことなどが示されています.
またEli Lillyの研究者らがLillyの社内ライブラリーにてPAINSフィルターを検証したところ,フィルター構造の多くは化合物の不安定性を現しているに過ぎないと示されました.
これ以上詳細に立ち入るのは本サイトの趣旨とは外れますので,興味のある方は以下の文献や記事を参照してみてください(全て英語です).
- 上述PubChemの論文:「Phantom PAINS: Problems with the Utility of Alerts for Pan-Assay INterference CompoundS」
- 上述Lillyの論文:「Investigating the Behavior of Published PAINS Alerts Using a Pharmaceutical Company Data Set」
- 上述のアメリカ化学会論説に対する反論:「Comment on The Ecstasy and Agony of Assay Interference Compounds」
- アメリカ化学会の方針に対するMolecular Designの記事:「PAINS and editorial policy」
- Lillyの論文に対するIN THE PIPLINEの記事:「PAINs Filters In the Real World」
- PAINS論文の著者による2017年の振り返り総説:「Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017–Utility and Limitations」
RDKitにおけるPAINSフィルターの実装
元文献では線形表記法の1つであるSLN形式でリストが提示されていましたので,RDKitではそれをより一般的なSMARTS形式に翻訳した上で実装されています.
開発初期段階ではRDKitにおける明示的な水素原子の取り扱いにやや問題があったために,本来よりもヒットする化合物数が少なくなるといったこともあったようですが,現在では改善されています.
・Changes in the 2016.03 Release: the FilterCatalog
・Curating the PAINS filters
・RDKit learns how to filter PAINS/BRENK/ZINC/NIH via FilterCatalog #536
フィルターカタログ
RDKitではPAINSフィルターはChem.rdfiltercatalogモジュールの一部として実装されています.PAINSの他にもZINCフィルターなどが実装されています.フィルターの種類は下記のコードのようにFilterCatalogs.namesでアクセス可能です.
from rdkit.Chem import FilterCatalog FilterCatalog.FilterCatalogParams.FilterCatalogs.names
{'PAINS_A': rdkit.Chem.rdfiltercatalog.FilterCatalogs.PAINS_A,
'PAINS_B': rdkit.Chem.rdfiltercatalog.FilterCatalogs.PAINS_B,
'PAINS_C': rdkit.Chem.rdfiltercatalog.FilterCatalogs.PAINS_C,
'PAINS': rdkit.Chem.rdfiltercatalog.FilterCatalogs.PAINS,
'BRENK': rdkit.Chem.rdfiltercatalog.FilterCatalogs.BRENK,
'NIH': rdkit.Chem.rdfiltercatalog.FilterCatalogs.NIH,
'ZINC': rdkit.Chem.rdfiltercatalog.FilterCatalogs.ZINC,
'ALL': rdkit.Chem.rdfiltercatalog.FilterCatalogs.ALL}
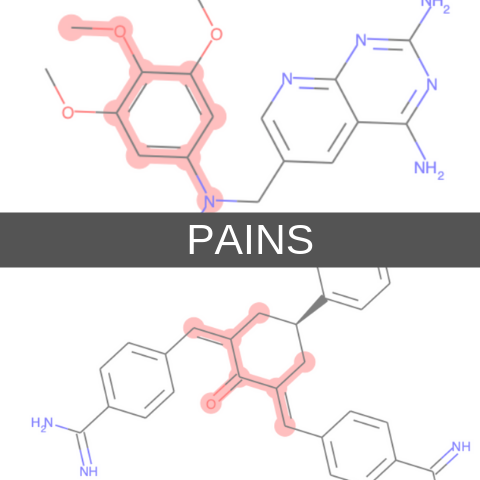
具体的にどのような構造がフィルタリング対象かを眺めるにはZINC15データベースのパターンを眺めるとよいと思います.下の画像ではPAINSフィルターの一部を取り出したものです.

フィルターカタログの具体的な使い方
フィルタリングの実行
Chem.rdfiltercatalog.FilterCatalog.GetMatches(mol)
具体的な使い方としては,まずChem.rdfiltercatalog.FilterCatalogParamsオブジェクトに先ほど見たフィルタリング要素を追加していき,望みのフィルターを作成します.コードとしては以下のようになります.
param = FilterCatalog.FilterCatalogParams() param.AddCatalog(FilterCatalog.FilterCatalogParams.FilterCatalogs.PAINS) filt = FilterCatalog.FilterCatalog(param)
続いて作成したフィルターに化合物を通していきます.HasMatchメソッドは該当構造の有無を,GetMatchesメソッドは該当構造全てをリストで返します.
それでは具体的に使い方を見ていきたいと思います.化合物としてはZINC15データベースから取得したある程度活性の高い化合物リストを用い,pandasのデータフレームとして読み込みます.
from rdkit import rdBase, Chem
from rdkit.Chem import AllChem, Draw, PandasTools
import pandas as pd
print(rdBase.rdkitVersion) # 2018.09.1
df = PandasTools.LoadSDF('./SDF/1uM.sdf.mol')
df.affinity = df.affinity.astype(float)
len(df) # 28372
df['PAINS'] = df.ROMol.map(filt.HasMatch)
df.head(2)
ここまでのデータフレームは以下のようになっています.

少し分布と統計量を見てみましょう.
import seaborn as sns sns.violinplot(x='PAINS', y='affinity', data=df) df.affinity.describe().round(2) df[ df.PAINS == False].affinity.describe().round(2) df[ df.PAINS == True].affinity.describe().round(2)

count 28372.00 mean 7.57 std 1.03 min 6.00 25% 6.71 50% 7.48 75% 8.30 max 11.00 Name: affinity, dtype: float64 count 26995.00 mean 7.58 std 1.02 min 6.00 25% 6.72 50% 7.49 75% 8.30 max 11.00 Name: affinity, dtype: float64 count 1377.00 mean 7.49 std 1.08 min 6.00 25% 6.55 50% 7.31 75% 8.19 max 10.68 Name: affinity, dtype: float64
特に大きな違いはなさそうです.続いて具体的にどのような構造がヒットしているかを見ていきます.
フィルターに一致した構造の取り扱い
GetMatchesメソッドは該当構造をChem.rdfiltercatalog.FilterCatalogEntryオブジェクトのリストで返します.
FilterCatalogEntryオブジェクトは
- description
- Reference
- Scope
というプロパティを有していて,どういった構造がヒットしたのかを知ることが可能です.
具体的なヒット構造のアトム番号を得るのはややトリッキーで,GetFilterMatchesメソッドを使うことになります.下のコードでは
- ヒット構造に関する説明を取得する関数を定義(descriptionプロパティ)
- ヒット構造のアトム番号のリストを返す関数を定義
- 定義した関数をデータフレームにmap関数で適用
- ランダムに選んだ6つの化合物に対して,ヒット構造をハイライトした状態で構造描画
という手順で進めています.またZINCデータベースでは分子は立体座標を持っていますので,2次元で見やすくなるように手を加えています.
def get_description(mol):
descriptions = []
if filt.HasMatch(mol):
catalog_entry_list = filt.GetMatches(mol)
for entry in catalog_entry_list:
descriptions.append(entry.GetProp('description'))
return descriptions
def get_matched_atoms(mol):
atom_pairs = []
if filt.HasMatch(mol):
catalog_entry_list = filt.GetMatches(mol)
for entry in catalog_entry_list:
pairs = [x[1] for x in entry.GetFilterMatches(mol)[0].atomPairs]
atom_pairs.append(pairs)
return atom_pairs
df['PAINS_desc'] = df.ROMol.map(get_description)
df['smiles'] = df.ROMol.map(Chem.MolToSmiles)
df['2D_mol'] = df.smiles.map(Chem.MolFromSmiles)
df['matched_atoms'] = df['2D_mol'].map(get_matched_atoms)
import numpy as np
random_list = list(np.random.choice(df[df.PAINS==True].index, size=6, replace=False))
Draw.MolsToGridImage(df['2D_mol'][random_list], subImgSize=(350,250),
highlightAtomLists=[x[0] for x in df['matched_atoms'][random_list]],
legends=[x[0] for x in df['PAINS_desc'][random_list]])

終わりに
今回は「RDKitのPAINSフィルターで化合物をスクリーニング」という話題について,
- PAINSフィルターとはなにか
- PAINSフィルターに関する議論
- RDKitにおけるフィルターカタログの実装
という内容を扱ってきました.PAINSフィルター自体は,必ずしもアッセイ系を阻害するような高頻度ヒット化合物のリストではないかもしれませんが,化合物の安定性などを考慮する際に忌避構造リストとして1つの指標になると思います.
コメント